机构名称:
¥ 1.0
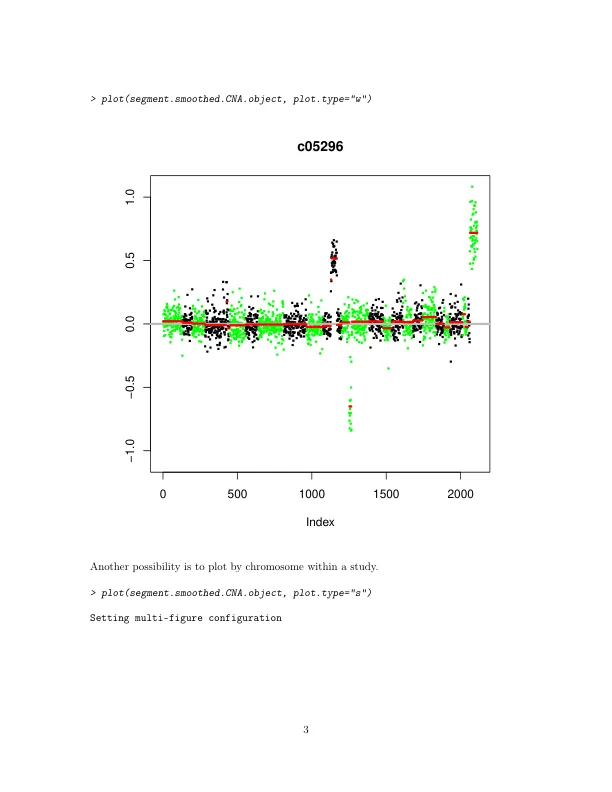
一个可能有趣但尚未提及的函数是 subset.CNA 。它允许通过染色体和样本对 CNA 对象进行子集设置,这样就不必对整个数据集进行分割。同样,subset.DNAcopy 允许对包含分割输出的 DNAcopy 对象进行子集设置。由于原始默认分割算法基于排列,因此需要 O(N2) 次计算,其中 N 是染色体上的标记数。新的默认算法要快得多。它包括一种混合方法,用于计算分割的 p 值,部分基于排列,部分基于高斯近似(在 1.2.0 之后的所有版本中可用),以及一条停止规则(在 1.5.0 之后的所有版本中可用),当有强有力的证据证明存在变化时宣布变化(Venkatraman 和 Olshen,2007)。我们不再建议对较大的数据集使用重叠窗口。仍然可以使用选项 p.method='perm' 运行完整的排列分析。如果新算法仍然太慢,可以使用参数 nperm(默认值为 10,000)减少混合方法中的排列数。但是,alpha(测试接受变化点的显著性水平)越低,所需的排列就越多。对于任何非默认值的 nperm 和 alpha 选择,都需要计算停止边界
DNAcopy:用于分析 DNA 拷贝数据的软件包
主要关键词




相关文件推荐