
机构名称:
¥ 4.0
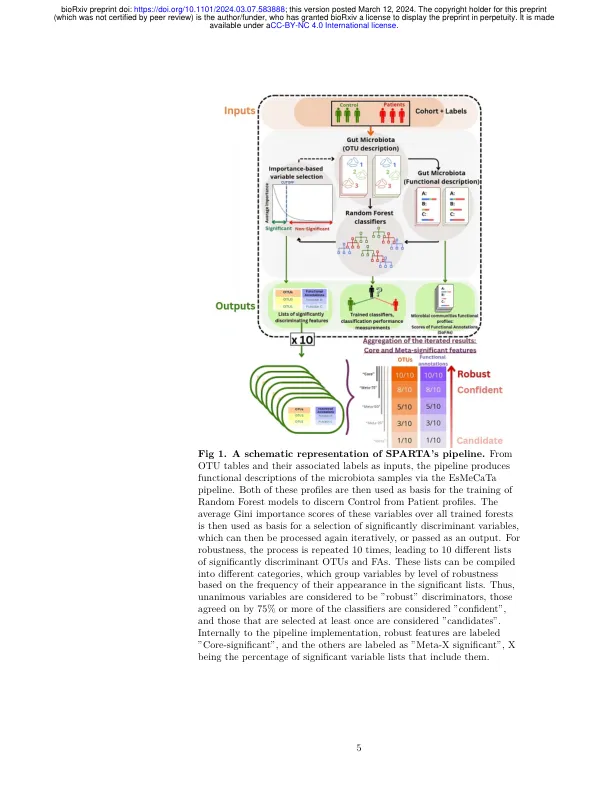
肠道微生物群的组成是各种疾病中的已知因素,事实证明是疾病状态自动分类的强大基础。需要在功能规模上更好地理解这个社区,因为这将增强这些APARACHES的生物解释性。在本文中,我们开发了一种计算管道,用于将肠道菌群的功能注释与自动分类过程相结合,并促进对其结果的下流解释。该过程作为输入分类组成数据(例如操作分类单元表(OTU)或Amplicon序列变体(ASV)丰度),并通过询问Uniprot数据库来将每个组合链接到其功能注释。肠道微生物群的功能性是由此基础构建的。二个pro纤维,微生物和功能性,用于训练随机的森林分类器,以辨别不健康的控制样品。然后根据可变的重要性进行自动选择,并且可以迭代该方法,直到分类性能降低为止。此过程表明,与微生物pro纤维相比,微生物群体转化为功能性纤维可比性,尽管表现略有下相比。通过重复,它还输出了一个强大的判别变量子集。这些选择比通过最先进的方法获得的选择更可靠,并且通过手动书目研究验证了其内容。还分析了选定的OTU和功能注释之间的互连,并揭示了重要的注释来自非选择OTU的累积影响。
与深色小胶质细胞相关的综合应力反应促进了小胶质细胞脂肪形成,并有助于神经变性RNA3DB:用于训练和基准的RNA结构预测的深度学习模型sparta:...
主要关键词




相关文件推荐





























