XiaoMi-AI文件搜索系统
World File Search SystemADCC

XMT-2056,一种指导的刺痛激动剂抗体-Drug ...
在这里,我们证明了XMT-2056表现出ADCC(抗体依赖性细胞介导的细胞毒性)功能,该功能随着STING途径的激活而协同,并诱导HER2表达癌细胞和FCγ-RIII +(CD16 + CD16 +)的HER2表达癌细胞中有效的癌细胞细胞活性。我们表明,XMT-2056和HT-19(未结合的父母抗HER2抗体)在FC效应的功能中保留了在PBMC共培养中以Fcγ-RI-Expectress Expectress Expectress Expectress的髓样细胞耗尽的PBMC共培养中的重要癌细胞杀死活性。通过Fcγ-RIII +免疫细胞的共排除,该活性消除了,这说明了XMT-2056的ADCC功能。在这种情况下,与HT-19相比,XMT-2056癌细胞杀伤活性显着增加,这表明刺痛激动剂有效载荷有助于对XMT-2056治疗观察到的差异活动。的确,癌细胞和免疫细胞与HT-19的共同培养和自由刺痛激动剂有效载荷共同培养增强了抗肿瘤反应,尽管其程度较小,而不是XMT-2056,这表明ADCC函数和Sting途径激活之间的协同作用。

化疗:药物 TZ 政策
Tafasitamab-cxix 是一种 Fc 修饰的单克隆抗体,可与表达于前 B 和成熟 B 淋巴细胞表面以及多种 B 细胞恶性肿瘤(包括弥漫性大 B 细胞淋巴瘤 (DLBCL))上的 CD19 抗原结合。与 CD19 结合后,tafasitamab-cxix 通过细胞凋亡和免疫效应机制介导 B 细胞裂解,包括抗体依赖性细胞毒性 (ADCC) 和抗体依赖性细胞吞噬作用 (ADCP)。在 DLBCL 肿瘤细胞中进行的体外研究中,与单独使用 tafasitamab-cxix 或来那度胺相比,tafasitamab-cxix 与来那度胺联合使用可提高 ADCC 活性。

增强自然杀伤细胞以克服癌症对 NK 细胞疗法的耐药性并增强抗体免疫疗法
自然杀伤 (NK) 细胞是先天免疫系统的细胞成分,可以识别和抑制癌细胞的增殖。NK 细胞可以通过直接裂解、分泌穿孔素和颗粒酶或通过抗体依赖性细胞介导的细胞毒性 (ADCC) 来消除癌细胞。ADCC 涉及 NK 细胞上的 Fc 伽马受体 IIIa (CD16) 与已经与癌细胞结合的抗体的恒定区结合。癌细胞使用多种机制来逃避 NK 细胞的抗肿瘤活性,包括抑制性细胞因子的积累、免疫抑制细胞(如髓源性抑制细胞 (MDSC) 和调节性 T 细胞 (Treg))的募集和扩增、NK 细胞受体的配体的调节。已经开发了几种策略来增强 NK 细胞的抗肿瘤活性,目的是克服癌细胞对 NK 细胞的抵抗力。改造和增强 NK 细胞毒性的三种主要策略包括使用调节性细胞因子增强 NK 细胞、过继性 NK 细胞疗法以及使用改造的 NK 细胞来增强基于抗体的免疫疗法。尽管前两种策略提高了基于 NK 细胞的疗法的疗效,但仍存在一些局限性,包括免疫相关不良事件、诱导免疫抑制细胞以及进一步产生对 NK 细胞杀伤的癌症抵抗力。克服这些问题的一种策略是结合介导 ADCC 的单克隆抗体 (mAb) 和具有增强抗癌活性的改造 NK 细胞。使用具有 ADCC 活性的 mAb 的优势在于它们可以激活 NK 细胞,但也有利于免疫效应细胞在肿瘤微环境 (TME) 中的积累。多项临床试验报告称,与单独使用 mAb 相比,将改造的 NK 细胞与具有 ADCC 活性的 mAb 相结合可以产生更好的临床反应。下一代临床试验采用工程化 NK 细胞和对 NK 细胞上表达的 CD16 具有更高亲和力的 mAb,将为癌症患者提供更有效、更高质量的治疗。

局部替诺福韦暴露前预防和粘膜 HIV ...
RV144 HIV 疫苗试验强调了包膜特异性非中和抗体 (nNAb) Fc 介导功能作为降低感染风险的免疫相关因素的重要性。由于暴露前预防 (PrEP) 和 HIV 疫苗被用作高危人群的联合预防策略,PrEP 对粘膜和全身 nNAb 功能的影响仍未确定。先前的动物和人体研究表明,使用 PrEP 后 HIV 血清转化后的 HIV 特异性抗体结合亲和力降低,这反过来可能会影响抗体的功能。在 CAPRISA 004 替诺福韦凝胶试验的血清转化者中,我们之前报告称,血浆和生殖道 (GT) 中 HIV 特异性结合抗体的检测和滴度明显更高,这使替诺福韦与安慰剂组有所区别。我们假设较高的 HIV 特异性抗体滴度和检测结果反映了相应增加的抗体依赖性中性粒细胞介导的吞噬作用 (ADNP) 和 NK 细胞激活的抗体依赖性细胞毒 (ADCC) 活性。在感染 HIV 后 3、6 和 12 个月,对 CAPRISA 004 替诺福韦凝胶试验中的 48 名血清转化者的 GT 和血浆样本中的 HIV 特异性 V1V2-gp70、gp120、gp41、p66 和 p24 抗体进行了 ADCP 和 ADCC 检测。在 6 个月和 12 个月时,替诺福韦组的 GT gp41 和 p24 特异性 ADNP 分别显著高于安慰剂组(p < 0.05)。替诺福韦组中血浆 gp120、gp41 和 p66 特异性 ADNP 以及 GT gp41 特异性 ADCC 随时间显著增加 (p < 0.05)。仅在替诺福韦组中,在感染后 6 个月,gp120 特异性 ADCC 与 gp120 抗体滴度 (r = − 0.54; p = 0.009) 之间以及 gp41 特异性 ADNP 与 gp41 特异性抗体滴度 (r = − 0.50; p = 0.015) 之间观察到显著的负相关性。此外,在替诺福韦组中,gp41 特异性 ADCC 与

抗CD20单克隆抗体概述
免疫系统相关的效应机制包括补体依赖性细胞毒性 (CDC) 和 Fcγ 受体 (FcγR) 介导的效应。FcγR 在几种免疫细胞上表达,例如中性粒细胞、巨噬细胞和自然杀伤 (NK) 细胞。通过 FcγR 的信号传导会触发抗体依赖性细胞介导的细胞毒性 (ADCC) 和细胞介导的吞噬作用 (ADCP)。11 补体的经典途径负责 CDC 活性,这是由于 C1q 和利妥昔单抗之间的结合。因此,该连接诱导膜攻击复合物的构成增加、调理作用引起的吞噬活性增强以及其他效应免疫成分的更多募集。12 在 mAb 通过 Fc 区与效应细胞 (FcγRIII) 相互作用后,ADCC 途径驱动由 NK 细胞介导的细胞毒性反应。活化的 NK 细胞通过膜通透性(释放穿孔素颗粒)和诱导程序性细胞死亡(通过颗粒酶 B 引发的 caspase 机制)导致靶细胞死亡。13,14 据报道,CDC 对 ADCC 机制可能产生不利影响,因为两者都竞争 mAb-CD20 复合物。体外研究表明利妥昔单抗具有更强的 CDC 活性;然而,体内模型报告称 ADCC 更有效。15 因此,CDC 对利妥昔单抗抗肿瘤作用的总体影响需要进一步的数据。最后,当 mAb 与巨噬细胞、单核细胞和中性粒细胞表面的其他 FcγR 相互作用时,就会发生 ADCP,从而导致靶细胞的吞噬。

介导的NK细胞的靶向靶向肿瘤... 与IRF2BPL基因变异的新型人类神经发育和神经退行性疾病 - 机械和治疗途径 leimusertib在临床前患者中具有抗肿瘤活性 - MACC1调节LGR5以促进结直肠癌的癌症干细胞特性 干细胞研究 研究文章NCBench:提供开放,可重复的,透明的, 癌症中与心血管健康相关的生活质量 干细胞研究 利用大型语言模型进行数据分析自动化 杂种免疫患者的后盘后状况的可能性;来自德国国家队列(Nako)的数据 评估基于机器学习的分类... 5-羟色胺转运蛋白依赖性组蛋白的血2-胎盘中有助于神经发育转录组 新型的色氨酸羟化酶抑制剂TPT-001逆转PAH,血管重塑和增殖 - 炎性基因表达 心脏发展和再生 - 多器官的努力 PRDM16突变确定性别特异性的心脏代谢,并识别两个新型的心脏代谢调节剂 早期小脑发育的调节 单细胞线粒体DNA突变中的伪影分析错误的系统发育推断 新型多动睡眠的分子演变
抽象背景可以通过特异性靶向触发抗体依赖性细胞介导的细胞毒性(ADCC)或通过遗传工程来表达嵌合抗原受体(CARS)来增强自然杀伤(NK)细胞的抗肿瘤活性。尽管抗体或汽车靶向,但某些肿瘤仍然对NK细胞攻击具有抗性。已知ICAM-1/LFA-1相互作用对NK细胞的自然细胞毒性的重要性,但它对ERBB2(HER2)特异性抗体曲妥珠单抗和ERBB2-培养基介导的NK细胞细胞毒性抗乳腺癌细胞诱导的ADCC的影响。方法,我们使用了表达高亲和力FC受体FcγRIIIA的NK-92细胞与曲妥珠单抗或ERBB2- CAR工程NK-92细胞(NK-92/5.28.Z)以及与ERBB2-CAR-2-CAR-2-CAR-2-CARID-ICAMID CYAMIS CYMINIC CYMINID CYMINIC CYMINID-CAR-2-CAR-2-CAR-92细胞(NK-92/5.28.z)结合使用,并或替代阻断NK细胞上的LFA-1。此外,我们特别刺激了FC受体,CAR和/或LFA-1,以研究其在免疫突触时的串扰,及其对抗体靶向抗体或靶向的NK细胞中脱粒和细胞内信号的贡献。结果阻断了LFA-1或ICAM-1的不存在会在曲妥珠单抗介导的ADCC中显着降低细胞杀伤和细胞因子释放,以针对ERBB2-阳性乳腺癌细胞,但在靶向汽车的NK细胞中并非如此。用5-Aza-2'-脱氧胞苷进行预处理,诱导ICAM-1上调,并反转ADCC中的NK细胞耐药性。此外,刺激抑制性NK细胞检查点NKG2A曲妥珠单抗单独没有充分激活NK细胞,需要额外的LFA-1共同刺激,而在CAR-NK细胞中ERBB2型车的激活会诱导的有效脱粒化,而与LFA-1无关。总内反射荧光单分子成像表明,CAR-NK细胞与排除ICAM-1的肿瘤细胞形成了不规则的免疫学突触,而曲妥珠单抗形成了典型的外周上分子超分子激活簇(PSMAC)结构。从机理上讲,ICAM-1的缺失不会影响ADCC期间的细胞 - 细胞粘附,而是导致通过PYK2和ERK1/2的信号降低,这是由CAR介导的靶向本质上提供的。

注射:药物政策-Medi -Cal
benralizumab是一种人源化的抗糖基化的单克隆抗体(IgG1,kappa),可直接与人白介素-5受体(IL-5Rα)的α亚基结合,分离常数为11 pm。IL-5受体在嗜酸性粒细胞和嗜碱性粒细胞的表面表达。在体外环境中,苯珠单抗FC结构域中缺乏岩藻糖,促进了与免疫效应细胞上的FCɣRIII受体的结合(45.5 nm),例如天然杀伤(NK)细胞,例如通过抗生素和嗜碱性细胞的细胞凋亡,通过抗体依赖性依赖性cyccccic cyccccic cytox cytoxcccccic cytoxcccccic cytoxcccccic cytox cytox cytoxcccccic cytox cytoxcccccic(炎症是哮喘发病机理中的重要组成部分。多种细胞类型(例如,肥大细胞,嗜酸性粒细胞,中性粒细胞,巨噬细胞,淋巴细胞)和介体(例如组胺,eicosanoids,白细胞素,细胞因子)涉及炎症。benralizumab通过与IL-5Rα链结合,通过ADCC降低嗜酸性粒细胞。然而,尚未确定哮喘中苯珠单抗作用的机制。

高亲和力 CD16 整合到 CRISPR/Cas9 编辑的 CD38 基因座中可增强原代人类自然杀伤细胞的 CD38 定向抗肿瘤活性
摘要 背景 过继转移具有增强的抗体依赖性细胞毒作用 (ADCC) 能力和对 CD38 靶向性抗性的自然杀伤 (NK) 细胞有可能增强达雷木单抗 (DARA) 的临床抗骨髓瘤活性。因此,我们试图开发一种有效的基于 CRISPR/Cas9 的基因编辑平台,以破坏离体扩增的 NK 细胞中的 CD38 表达 (CD38 敲除 (KO)),并同时为 CD38 KO NK 细胞配备高亲和力 CD16 (CD16-158V) 受体。方法 使用 Cas9 核糖核蛋白复合物生成 CD38 KO 人 NK 细胞。通过结合信使 RNA (mRNA) 转染 CD38 KO NK 细胞和在 CD38 位点插入靶向基因以介导基因敲入 (KI),扩展了该平台。在体外和 MM.1S 异种移植小鼠模型中测试了这些基因编辑的 NK 细胞在 DARA 存在下持续存在和介导 ADCC 的能力。结果在体外扩增的 NK 细胞中实现了高效的 CD38 基因破坏,而不会影响其增殖或功能能力。CD38 KO 赋予了对 DARA 诱导的 NK 细胞自相残杀的抗性,在体外和 MM.1S 异种移植小鼠模型中,在 DARA 存在下,能够持续存在并增强对骨髓瘤细胞系的 ADCC。CD38 KO NK 细胞可以通过转染编码 CD16-158V 受体的 mRNA 进一步修饰,从而增强 DARA 介导的 ADCC。最后,我们观察到针对 CD38 基因座的同源定向修复模板促进了有效的 2 合 1 CD38 KO 与截短 CD34 报告基因和 CD16-158V 受体的 KI 结合,CD38 KO /CD16 KI NK 细胞在体外和体内均表现出 DARA 介导的 ADCC 的进一步增强。结论使用体外扩增的 CD38 KO /CD16 KI NK 细胞进行过继免疫治疗有可能提高 DARA 的临床疗效。通过将互补的基因工程策略整合到 CD38 KO 制造平台中,我们生成了具有显著增强的 CD38 定向抗肿瘤活性的 NK 细胞,为在临床上探索这种免疫治疗策略奠定了坚实的基础。
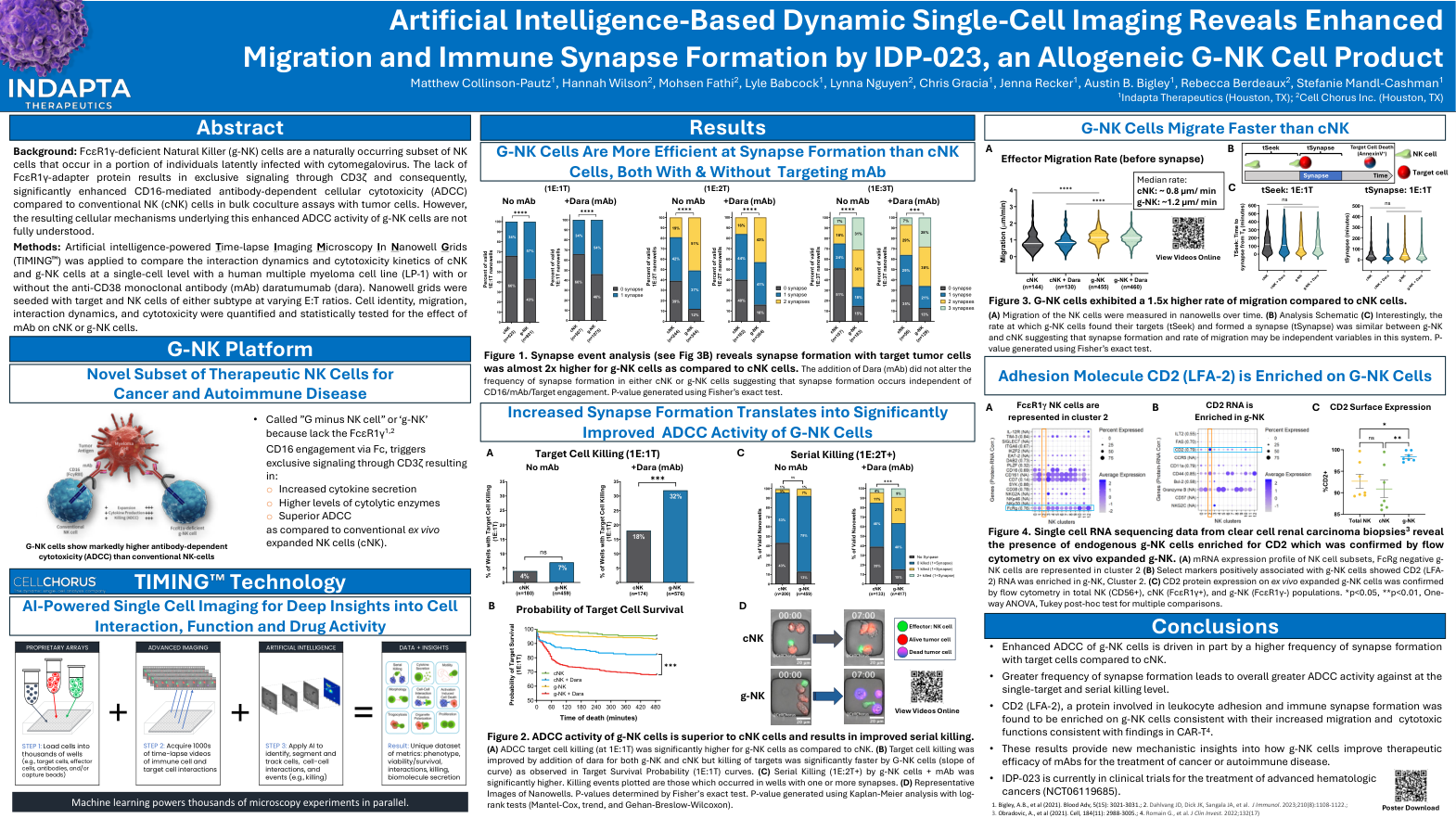
基于人工智能的动态单细胞成像揭示同种异体 G-NK 细胞产品 IDP-023 增强的迁移和免疫突触形成
图 2。g-NK 细胞的 ADCC 活性优于 cNK 细胞,并可改善连续杀伤。(A) 与 cNK 相比,g-NK 细胞的 ADCC 靶细胞杀伤率(1E:1T)明显更高。(B) 通过添加 dara,g-NK 和 cNK 的靶细胞杀伤率均有所提高,但如靶标存活概率 (1E:1T) 曲线所示,G-NK 细胞的靶标杀伤速度明显更快(曲线斜率)。(C) g-NK 细胞 + mAb 的连续杀伤率(1E:2T+)明显更高。绘制的杀伤事件发生在具有一个或多个突触的孔中。(D) 纳米孔的代表性图像。P 值由 Fisher 精确检验确定。使用 Kaplan-Meier 分析和对数秩检验 (Mantel-Cox、趋势和 Gehan-Breslow-Wilcoxon) 生成 P 值。

CAR介导的NK细胞的靶向克服了由ICAM-1下调引起的肿瘤免疫逃逸 种系遗传变异与免疫检查点抑制剂治疗的癌症患者的胰岛素依赖性糖尿病的发展有关 使用嵌合内吞式受体对CAR T细胞活性的敏感操纵 图S1。用于细胞内染色的流式细胞术的门控策略。 的百分比 肿瘤相关的巨噬细胞:癌症的潜在治疗策略和未来前景 在奥地利,瑞士和德国中心的黑色素瘤患者中,在黑色素瘤患者中使用Talimogene laherparepvec(T-VEC) ... 患者的现有TGF-β特异性T细胞免疫 免疫检查点抑制剂治疗和动脉粥样硬化心血管疾病:新兴的临床问题 非中的免疫细胞浸润模式 携带EGFR突变的小细胞肺癌患者 口服SMEDD促进绿素酸的淋巴运输和肠系膜淋巴结靶标,可有效T细胞抗肿瘤免疫 e002954.full.pdf 术前Sitravatinib和Nivolumab在口腔癌中的抗肿瘤免疫作用:大雪窗口研究 双特异性抗体的免疫原性评估 免疫肿瘤趋势:临床前模型,生物标志物和临床开发 多发性骨髓瘤中与肿瘤相关的巨噬细胞 tipe2缺失提高了采用转移的NK细胞的治疗潜力
抽象背景可以通过特异性靶向触发抗体依赖性细胞介导的细胞毒性(ADCC)或通过遗传工程来表达嵌合抗原受体(CARS)来增强自然杀伤(NK)细胞的抗肿瘤活性。尽管抗体或汽车靶向,但某些肿瘤仍然对NK细胞攻击具有抗性。已知ICAM-1/LFA-1相互作用对NK细胞的自然细胞毒性的重要性,但它对ERBB2(HER2)特异性抗体曲妥珠单抗和ERBB2-培养基介导的NK细胞细胞毒性抗乳腺癌细胞诱导的ADCC的影响。方法,我们使用了表达高亲和力FC受体FcγRIIIA的NK-92细胞与曲妥珠单抗或ERBB2- CAR工程NK-92细胞(NK-92/5.28.Z)以及与ERBB2-CAR-2-CAR-2-CAR-2-CARID-ICAMID CYAMIS CYMINIC CYMINID CYMINIC CYMINID-CAR-2-CAR-2-CAR-92细胞(NK-92/5.28.z)结合使用,并或替代阻断NK细胞上的LFA-1。此外,我们特别刺激了FC受体,CAR和/或LFA-1,以研究其在免疫突触时的串扰,及其对抗体靶向抗体或靶向的NK细胞中脱粒和细胞内信号的贡献。结果阻断了LFA-1或ICAM-1的不存在会在曲妥珠单抗介导的ADCC中显着降低细胞杀伤和细胞因子释放,以针对ERBB2-阳性乳腺癌细胞,但在靶向汽车的NK细胞中并非如此。用5-Aza-2'-脱氧胞苷进行预处理,诱导ICAM-1上调,并反转ADCC中的NK细胞耐药性。此外,刺激抑制性NK细胞检查点NKG2A曲妥珠单抗单独没有充分激活NK细胞,需要额外的LFA-1共同刺激,而在CAR-NK细胞中ERBB2型车的激活会诱导的有效脱粒化,而与LFA-1无关。总内反射荧光单分子成像表明,CAR-NK细胞与排除ICAM-1的肿瘤细胞形成了不规则的免疫学突触,而曲妥珠单抗形成了典型的外周上分子超分子激活簇(PSMAC)结构。从机理上讲,ICAM-1的缺失不会影响ADCC期间的细胞 - 细胞粘附,而是导致通过PYK2和ERK1/2的信号降低,这是由CAR介导的靶向本质上提供的。