XiaoMi-AI文件搜索系统
World File Search SystemHISEQ
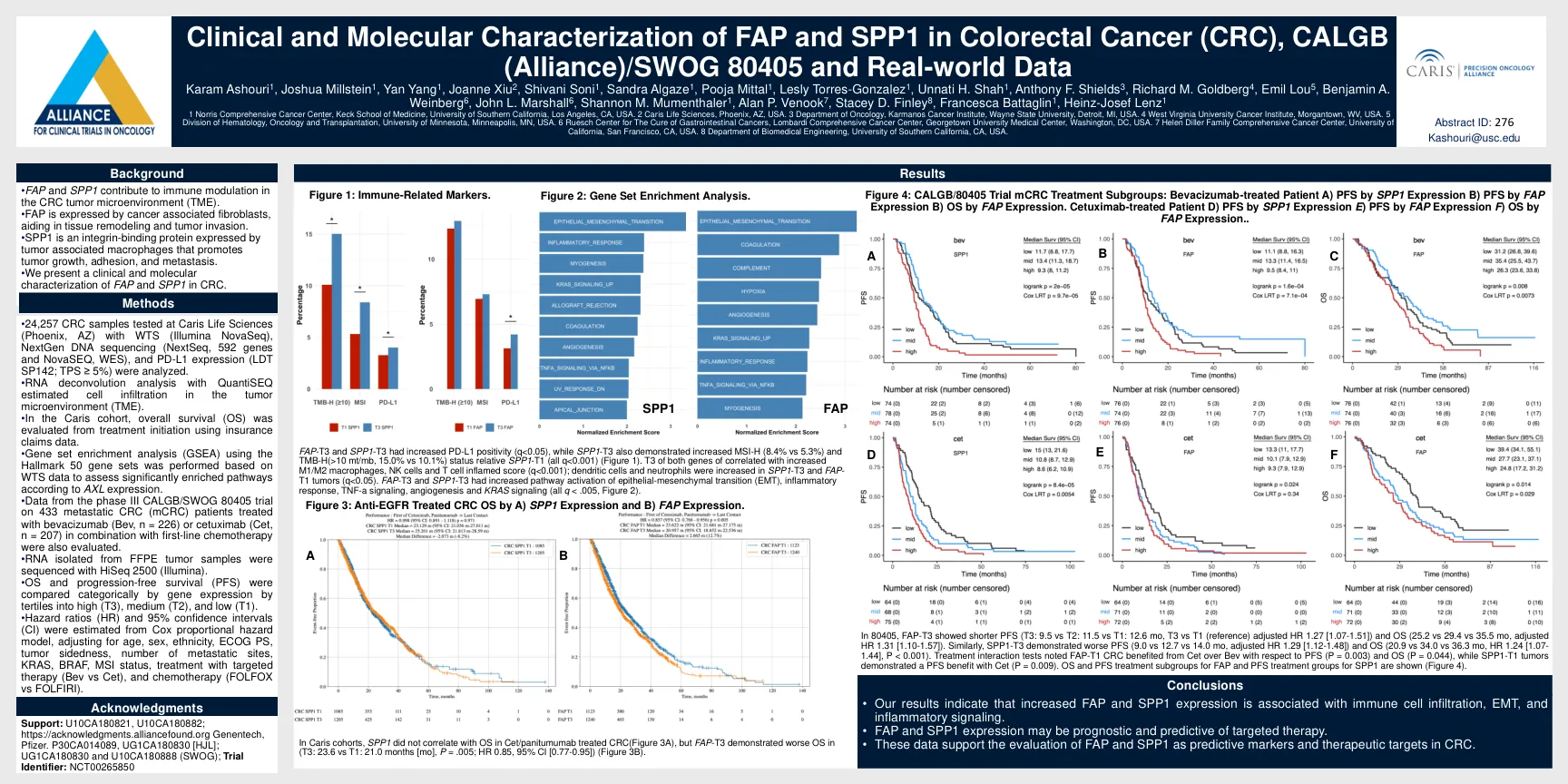
FAP 和 SPP1 的临床和分子表征...
• 分析了 Caris Life Sciences(亚利桑那州凤凰城)使用 WTS(Illumina NovaSeq)、NextGen DNA 测序(NextSeq、592 个基因和 NovaSEQ、WES)和 PD-L1 表达(LDT SP142;TPS ≥ 5%)测试的 24,257 个 CRC 样本。• 使用 QuantiSEQ 进行 RNA 反卷积分析估计肿瘤微环境 (TME) 中的细胞浸润。• 在 Caris 队列中,使用保险索赔数据从治疗开始评估总体生存率 (OS)。• 根据 WTS 数据使用 Hallmark 50 基因组进行基因集富集分析 (GSEA),以根据 AXL 表达评估显著富集的通路。 • 还评估了 III 期 CALGB/SWOG 80405 试验的数据,该试验涉及 433 名接受贝伐单抗 (Bev,n = 226) 或西妥昔单抗 (Cet,n = 207) 联合一线化疗治疗的转移性 CRC (mCRC) 患者。 • 使用 HiSeq 2500 (Illumina) 对从 FFPE 肿瘤样本中分离的 RNA 进行测序。 • 根据基因表达将 OS 和无进展生存期 (PFS) 分为高 (T3)、中 (T2) 和低 (T1) 三分位数进行分类比较。 • 根据 Cox 比例风险模型估计风险比 (HR) 和 95% 置信区间 (CI),并根据年龄、性别、种族、ECOG PS、肿瘤单侧性、转移部位数量、KRAS、BRAF、MSI 状态、靶向治疗 (Bev vs Cet) 和化疗 (FOLFOX vs FOLFIRI) 进行调整。

CSPP1,TMEM67,PLP1和GAN相关的突变...
背景和目标:神经系统疾病严重影响患者的心理,性格和运动功能,全球患病率上升,尤其是在低收入和中等收入的国家。这项研究旨在评估儿科神经系统疾病中的基因突变,使患者有助于我们对这些疾病的遗传基础的理解。方法:在当前的调查中,所有在2023年至2024年期间被转诊至神经病学部门的母体迹象的患者均已评估。使用Agilent Sureselect Human All Exon Kit V6富含来自患者的DNA样品,然后根据制造商的程序在Illumina HISEQ 4000平台上进行了测序。结果:在当前的横断面研究中,评估了13例母体神经系统疾病患者,包括6名男性(46%)和7个女性(54%)。我们的结果确定了遗传性神经系统疾病,包括乔伯特综合征,Pelizaeus-Merzbacher病和巨型轴突神经病1。我们的数据在PLP1基因的外显子8(NM_001128834.3:c.772a> c; p.met258leu)中鉴定出了一个新的错义突变,并在患有pelizaeus-merzbacher病的患者中具有X连锁的隐性遗传。基因变体,包括外显子20(NM_001382391.1:c.2259_2260delaa; P.Glu7555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555GLYFSTER30)和常染色体隐性膜体固体TMEM67在Exon 8(Exon 8(NM_1537)中> C.725)在乔伯特综合征患者中检测到p.asn242ser)。最后,在患有巨大轴突神经病1的患者中,检测到纯合gan突变(NM_022041.4:c.1177t> c; p.cys393arg)。结论:我们的发现对于理解神经系统疾病的病理生理可能很有用。此外,这项研究还表明了遗传分析在使用神经系统疾病患者中使用治疗策略的重要性。

补充材料
手术获得的原发性肿瘤组织样品和相应的连续血样品,分别获得了体细胞突变筛查和ctDNA监测。使用三组结直肠癌患者(CRC)的三个不同平台对原发性肿瘤进行了测序分析。分别分析了11、27和14例患者的肿瘤样本,分别为1、2和3组。使用Clearseq综合癌症小组(Agilent Technologies,Inc.,Santa Clara,CA)使用Illumina HISEQ 2000 Sequencer(Illumina,Inc。,San Diego,CA)分析集1,该基因针对151个疾病相关的基因(Maintext的参考文献34)。 分别使用ION Proton™和ION S5™系统(Thermo Fisher Scientific,Waltham,MA)分析了集2和3,其定制面板的靶向39个基因,这些基因经常在CRC中改变。 1集1样品在我们的先前研究中进行了分析,该研究考虑了原发性肿瘤的三个区域以及外周血单核细胞(PBMCS)(总共四个样品)的相应DNA,以评估肿瘤内遗传异质性对循环肿瘤DNA DNA(CTDNA)(CTDNA)(参考文本34)的影响。 在该研究中,结果表明,肿瘤遗传异质性并不是CTDNA分析的主要障碍,前提是从肿瘤的单个区域中选择具有足够变异等位基因频率(VAF)的体突变。 本研究中原发性肿瘤测序的最高优先级是检测一些具有高VAF的体细胞突变。集1,该基因针对151个疾病相关的基因(Maintext的参考文献34)。集2和3,其定制面板的靶向39个基因,这些基因经常在CRC中改变。1集1样品在我们的先前研究中进行了分析,该研究考虑了原发性肿瘤的三个区域以及外周血单核细胞(PBMCS)(总共四个样品)的相应DNA,以评估肿瘤内遗传异质性对循环肿瘤DNA DNA(CTDNA)(CTDNA)(参考文本34)的影响。在该研究中,结果表明,肿瘤遗传异质性并不是CTDNA分析的主要障碍,前提是从肿瘤的单个区域中选择具有足够变异等位基因频率(VAF)的体突变。本研究中原发性肿瘤测序的最高优先级是检测一些具有高VAF的体细胞突变。此外,三个肿瘤区域中通常检测到的突变仅限于一组基因,其中包括TP53,APC,KRAS,PIK3CA,FBXW7和BRAF。

厌氧硝酸盐还原的基因组草案,苯
Thermincola Mag的组装使用了多个先前报道的数据集(6)。Illumina配对端(NCBI登录:SRR24043423)和Mate-pair(NCBI登录:SRR24043417)读数是从2013年从称为NRBC亚养殖Cartcons19获得的。配对末端的读数进行了测序,并使用Nextera Mate Pair库制剂制备套件对配偶对读数进行了测序。使用Trimmomaticv。0.32(7)处理所有原始读数,然后使用Abyssv。1.3.7(8),以创建与All-Paths-LGv。4.7.0(9)中生成的脚手架合并的Unitigs,使用gap填充Perl Script(10)基于Tang S1中的script in Dang et et eT eT eT eT eT eT eT eT script。(11)。由于该元基因组组装中的不确定核苷酸数量大量(JARXNP010000000),因此采取了进一步的步骤。在2018年,使用HISEQ PE群集Kit v4 cbot(Illumina)对NRBC亚培养(FES-DIASIS)进行了测序,没有其他质量控制措施(NCBI登录:SRR24043422)使用IDBAv。1.1.1.1(12)(12)和BINNENNNEND和VINNEND。在157个重叠群(NCBI登录:Javsmv000000000.1)中分配给Thermincola的垃圾箱如前所述(6)。将这157个重叠群纳入上述深渊/全paths-lg间隙填充工作流程中,生成了一个26 contig组件,该组件是通过使用BBMAPv。38.94(14)来策划映射来解决歧义的26 contig组件。读取映射可视化是使用Geneiousv。8.1.8进行的,并使用NCBI的原始基因组注释进行了基因组注释Finally, long reads from a 2020 NRBC subculture called 10L-NRBC, sequenced according to the manufac turer's instructions using PacBio RSII with the SMRTbell Express Template Prep Kit 2.0 ( SRR24043419 ) without shearing or size selection (Pacific Biosciences), were used to join adjacent contigs using the de novo assembly tool in Geneious v. 8.1.8(15),导致20碳组装。

鉴定人类细胞中复制应力反应的关键因素Louise M.E. Janssen 1,Empar Baltasar Perez 1,Chantal Vaartin
方法在补充了10%FCS,1%谷歌补充剂(Gibco),100 U/ml青霉素和100μg/ml链霉菌素的IMDM(Gibco)中培养了衍生成近单倍型HAP1细胞的细胞培养。siRNA转染是根据制造商的指南使用Rnaimax(Invitrogen)进行的。在这项研究中使用了以下siRNA:Sinon-targetable(Dharmacon),Sipolg2(地平线,TargetPlus,SmartPool),SIMRPL23(Horizon,Targetplus,TargetPlus,Smartpool)。将所有药物(Aphidicolin,Hu,Olaparib,Rad51i(B02),DNA-PKI(NU74441)和寡霉素A)溶解在DMSO中,并以指示浓度使用。细胞使用具有137CS源的γ提取器(最佳疗法)进行γ辐射。生长测定HAP1细胞以1500个细胞/孔的密度将HAP1细胞铺在96孔板中,并被视为5天。5天后,使用100%甲醇固定细胞,并在室温下使用Crystal Violet染色2H。随后,将晶体紫溶解在10%乙酸中,并使用Biotek Epoch Epoch分光光度计在595 nm处测量强度。使用非线性拟合,sigmoidal,4pl,x是log(浓度),将这些测量值用于棱镜中的IC50计算。在9mm玻璃盖上生长免疫荧光细胞,并在室温下以4%甲醛和0.2%Triton X-100固定10分钟。使用了以下抗体:人类抗克雷斯特(Cortex Biochem,CS1058),兔抗PH3SER10(Campro,#07-081),小鼠抗ERCC6L(PICH)(ABNOVA,ABNOVA,000548421-B01P)。所有初级抗体在4°C的夜间孵育。使用固定缓冲液I(BD生物科学)固定细胞。细胞。二级抗体(分子探针,Invitrogen)和DAPI在室温下孵育2小时。使用延长金(Invitrogen)安装盖玻片。使用具有60倍1.40 Na油目标的Deltavision Deonvolution显微镜(Applied Precision)获取图像。SoftWorx(应用精度),ImageJ,Adobe Photoshop和Illustrator CS6用于处理获得的图像。单倍体插入诱变筛选基因对用APH或HU处理的HAP1细胞的存活至关重要,如先前所述35,使用单倍体插入诱变筛查鉴定。诱变的HAP1细胞是从Brummelkamp实验室获得的。简短地,获得HAP1细胞的诱变如下:在HEK293T细胞中产生了基因陷阱逆转录病毒。每天两次收获逆转录病毒至少三天,并通过离心(使用SW28转子进行2小时,21,000 rpm,4°C,4°C)进行沉淀。在8μg/ml硫酸素硫酸素的存在下,在T175烧瓶中至少连续两天,在8μg/ml硫酸素的存在下,将大约4000万个HAP1细胞通过浓缩基因陷阱病毒的转导而被诱变。在包含10%DMSO和10%FCS的IMDM培养基中冷冻诱变细胞。解冻后,在存在27.5 nm adphidicolin或100μmHu的情况下,将诱变的HAP1细胞转移了10天。传递后,通过胰蛋白酶-EDTA收集细胞,然后进行沉淀。为了最大程度地减少潜在地含有杂合突变的二倍体细胞的混杂,用DAPI染色固定的细胞,以允许使用Astrios Moflo对G1单倍体DNA含量进行分类。将3000万个排序的细胞在56°C下裂解过夜,以使使用DNA迷你试剂盒(QIAGEN)进行基因组DNA分离。插入位点映射基因陷阱插入位点通过LAM-PCR放大,然后进行捕获,ssDNA接头连接和指数放大,并在测序之前使用含有Illumina适配器的引物,如前所述,如前所述35。映射和插入位点的分析以前描述了78。简短地,在对HISEQ 2000或HISEQ 2500(Illumina)进行测序之后,将插入位点映射到人类基因组(H19),允许一个不匹配,并与RefSeq坐标相交,以将插入位点分配给基因。基因区域在相对链上重叠的基因区域没有考虑进行分析,而对于在相同链基因名称上重叠的基因是串联的。对于每种复制和两种药物治疗(APH或HU)基因的必要性都是通过二项式检验确定的。合成致死性。一个基因通过所有Fisher的测试,其p值截止为0.05,效应大小至少为0.12(减法比率wt sense比率 - 复制应力条件感官比率)。

MSAKEN,突尼斯:一个普通的父亲祖先确认...
突尼斯公民科学家技术总监。摘要MSAKEN CITY(突尼斯)被认为是由五名从西亚迁移的相关男子在公元1360年左右建立的。人口将随着这些创始人的后代以及来自突尼斯不同地区的其他人口的到来而发展。为了阐明创始人人口的TMRCA并揭示其地理起源,使用商业公司的服务检查了来自MSAKEN不同家族的23名男性,使用NG测试技术检查了12至440 Y的染色体短串联重复序(STR)和单核苷酸多态性(SNP)标记。八个样品被基因分型用于SNP,以确定其单倍群。为了完善系统发育,在一个样品上以300,000 bp的速度进行了传统的Sanger测试(在Y Chr中步行中)。还使用下一代测试(BIGY)测试了七个样本(BIGY),其中涵盖了2000万bp的Y染色体,重叠了85%的黄金标准区域(通过YCC将其放置在系统发育树上的染色体Y位置),使用NGS Instruments,Hiseq 2000和2500和2500和2500。使用SQL脚本和数据挖掘工具,对STR结果与来自不同来源和数据库的数据进行了比较,以查找匹配的单倍型。发现所有STR结果的每12个标记不超过三个不匹配,每67个标记不超过6个不匹配,而SNP结果表明,所有测试样品均属于其子组中的单倍群J-M172。依靠通用的Str标记值,我们定义了MSAKEN-HAPLOTYPE。ng测试我们的样品以及添加到yfull.com树中的测试使我们能够完善J-L24的系统发育,并发现样品均属于J-L271单倍群,共享54个独家SNP。基于NG测试的最新共同祖先(TMRCA)的计算时间,范围为1500至6200 YBP,显示出约5400 YBP的强瓶颈。收集结果的变化显示了东安纳托利亚,当今亚美尼亚,阿塞拜疆和西伊朗的J-L192的地理根。属于J-M172(J2)单倍群的随机突尼斯STR单倍型的20%至30%表现出MSAKEN-HAPLOTYPE。关键词:MSAKEN DNA;突尼斯DNA; salar dna;中国DNA;单倍群J2

交叉
单分子实时 (SMRT) DNA 测序技术 (Pacific Biosciences) 生成的长读段是高质量叶绿体 (1, 2) 和线粒体基因组序列组装的起点之一。栽培的葡萄树 Vitis vinifera 极易受到病原体的感染。抗性品种如种间杂交品种‘Börner’ (V. riparia GM183 [母株] V. cinerea Arnold [花粉供体]) 被用作培育优良葡萄品种的砧木。我们从 SMRT 读段中组装并注释了‘Börner’的叶绿体 (cp_Boe) 和线粒体 (mt_Boe) 基因组序列。除非另有说明,所有生物信息学工具均采用默认参数。从品种“Börner”的幼叶中提取基因组 DNA(3),并在 Sequel I 测序仪(1Mv3 SMRT 细胞、结合试剂盒 v3.0、测序化学 v3.0,均来自 PacBio)上进行测序。通过 BLASTN(BLAST 2.7.1)搜索(4)筛选质体或线粒体序列(RefSeq 版本 91),筛选出潜在的质体或线粒体读段。使用的标准如下:读段长度,500 个核苷酸(nt)以上;同一性,70% 以上;查询覆盖率,30% 以上。 292,574 个潜在质体读段(共 2,715,983,671 nt;N50,12,829 nt)和 426,918 个潜在线粒体读段(3,928,350,102 nt;N50,12,624 nt)分别用 Canu v1.7(5)进行组装。每个最长的重叠群都与 V. vinifera 的叶绿体(6)或线粒体(7)基因组序列具有高度相似性。随后,使用 Bandage(8)确认组装正确。手动修剪环状基因组中重叠的末端序列,并将起始序列与葡萄参考序列比对。用 Arrow(SMRT Link 版本 5.1.0.26412)对组装体进行三次完善。最后一轮精炼将起始点移至序列的相反位置。为了帮助注释,根据制造商的说明,使用 peqGOLD 植物 RNA 试剂盒 (Peqlab) 从“Börner”组织中提取 RNA。根据 TruSeq RNA 样品制备 v2 指南,从 1,000 ng 总 RNA 制备索引 Illumina 测序文库。将得到的转录组测序 (RNA-Seq) 文库以等摩尔量汇集,并在 HiSeq 1500 仪器上以 2 100-nt 双端格式进行测序。cp_Boe (161,008 bp;GC 含量,37.4%) 和 mt_Boe (755,068 bp;GC 含量,44.3%) 使用 Web 服务 GeSeq v1.66 进行注释(cp_Boe 的具体设置:

全基因组测序和分析 - Illumina
全基因组测序和分析 - 基于 Illumina Rhabdoid (RT) Illumina 基因组板的文库构建(350-450bp 插入大小):将 96 孔格式的 2ug 基因组 DNA 通过 Covaris E210 超声处理 30 秒进行碎裂,使用 20% 的“占空比”和 5 的“强度”。双端测序文库是按照 BC 癌症机构基因组科学中心 96 孔基因组 ~350bp-450bp 插入 Illumina 文库构建协议在 Biomek FX 机器人(Beckman-Coulter,美国)上准备的。简单来说,DNA在96孔微量滴定板中用Ampure XP SPRI 珠子纯化(每60uL DNA 40-45uL 珠子),在单一反应中分别用T4 DNA聚合酶、Klenow DNA聚合酶和T4多核苷酸激酶进行末端修复和磷酸化,然后用Ampure XP SPRI 珠子进行清理,并用Klenow片段(3'到5'外显子减去)进行3' A加尾。用Ampure XP SPRI 珠子清理后,进行picogreen定量以确定下一步接头连接反应中使用的Illumina PE接头的数量。使用 Ampure XP SPRI 珠子纯化接头连接产物,然后使用 Illumina 的 PE 索引引物组,用 Phusion DNA 聚合酶(美国赛默飞世尔科技公司)进行 PCR 扩增,循环条件为:98˚C 30 秒,然后 6 个循环,98˚C 15 秒,62˚C 30 秒,72˚C 30 秒,最后在 72˚C 延伸 5 分钟。使用 Ampure XP SPRI 珠子纯化 PCR 产物,并使用高灵敏度分析(美国珀金埃尔默公司)用 Caliper LabChip GX 检查 DNA 样本。所需大小范围的 PCR 产物经过凝胶纯化(在内部定制机器人中使用 8% PAGE 或 1.5% Metaphor 琼脂糖),并使用 Agilent DNA 1000 系列 II 检测和 Quant-iT dsDNA HS 检测试剂盒使用 Qubit 荧光计(Invitrogen)评估和量化 DNA 质量,然后稀释至 8nM。在使用 v3 化学法在 Illumina HiSeq 2000/2500 平台上生成 100bp 配对末端读数之前,通过 Quant-iT dsDNA HS 检测确认最终浓度。全基因组亚硫酸盐-Seq 文库构建和测序:使用 1-5 mg Qubit(Life Technologies,加利福尼亚州卡尔斯巴德)定量基因组 DNA 进行文库构建,如所述(Gascard 等人,2015 年)。为了追踪亚硫酸盐转化的效率,将 1 ng 未甲基化的 lambda DNA (Promega) 掺入使用 Qubit 荧光法定量的 1 µg 基因组 DNA 中,并排列在 96 孔微量滴定板中。使用 Covaris 超声处理将 DNA 剪切至 300 bp 的目标大小,并使用 DNA 连接酶和 dNTP 在 30o C 下对片段进行末端修复 30 分钟。使用 2:1 AMPure XP 珠子与样品比例纯化修复后的 DNA,并在 40 µL 洗脱缓冲液中洗脱以准备 A 尾;这涉及使用 Klenow 片段和 dATP 将腺苷添加到 DNA 片段的 3' 端,然后在 37o C 下孵育 30 分钟。用磁珠清理反应后,将胞嘧啶甲基化双端接头(5'-AmCAmCTmCTTTmCmCmCTAmCAmCGAmCGmCTmCTTmCmCGATmCT-3' 和 3'-GAGmCmCGTAAGGAmCGAmCTTGGmCGAGAAGGmCTAG-5')在 30oC 下连接到 DNA 20 分钟,并纯化接头两侧的 DNA 片段珠。在亚硫酸盐转化之前,用 10 个 PCR 循环扩增一份文库片段,并在 Agilent Bioanalyzer 高灵敏度 DNA 芯片上进行大小测定。扩增子的长度在 200-700 bp 之间。使用 EZ Methylation-Gold 试剂盒(Zymo Research)按照制造商的方案实现甲基化接头连接的 DNA 片段的亚硫酸盐转化。五次循环