XiaoMi-AI文件搜索系统
World File Search SystemTRAE

简介
ADC,抗体且药物结合; Af -hpa,auristatin f羟丙基酰胺; AST,天冬氨酸氨基转移酶; CR,完全响应; DAR,药物与抗体比; DCR,疾病控制率; DL,剂量水平; DOR,响应持续时间; ECOG PS,东方合作肿瘤学组绩效状况; Fcγ,可结晶的伽马受体; G,等级; IHC,免疫组织化学; ILD,间质性肺疾病; iv,静脉注射; MTD,最大耐受剂量; NAPI2B,依赖钠的磷酸转运蛋白2B; NSCLC,非小细胞肺癌; OC,卵巢癌; ORR,客观响应率; PARP,聚ADP核糖聚合酶; PDX,患者衍生的异种移植物; PK,药代动力学; QW,每周;恢复,实体瘤的反应评估标准; RP2D,建议的2期剂量; SD,稳定疾病; SEM,平均值的标准误差; TRAE,与治疗相关的不良事件;紫外线,紫外线。
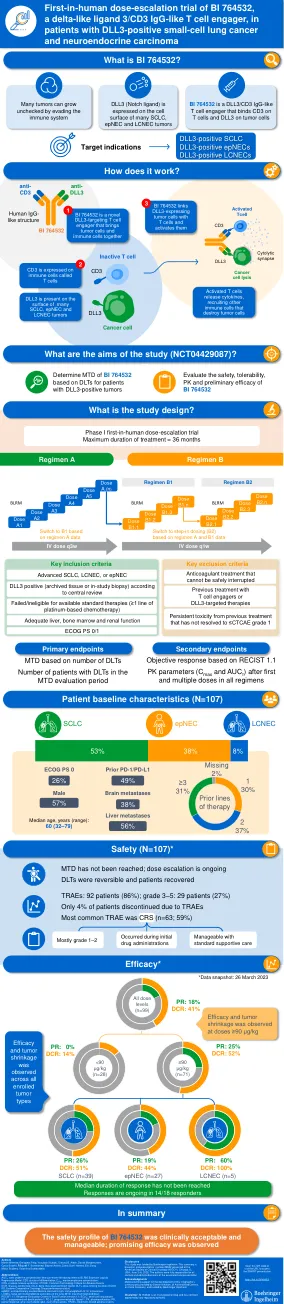
BI 764532 的首次人体剂量递增试验,delta-...
缩写 AUC τ ,给药间隔内浓度-时间曲线下面积;BLRM,贝叶斯逻辑回归模型;CD3,分化簇;C max ,最高血浆浓度;CRS,细胞因子释放综合征;CTCAE,不良事件常用术语标准;DCR,疾病控制率;DLL3,delta 样典型 Notch 配体;DLTs,剂量限制性毒性;ECOG PS,东部肿瘤协作组体能状态;epNEC,肺外神经内分泌癌;IgG,免疫球蛋白 G;IV,静脉内;LCNEC,肺大细胞神经内分泌癌;MTD,最大耐受剂量;RECIST 1.1,实体肿瘤疗效评价标准 1.1 版;SCLC,小细胞肺癌;PD-1,程序性细胞死亡蛋白-1;PD-L1,程序性死亡配体-1; PK,药代动力学;PR,部分缓解;q1w,每周;q3w,每三周;TRAE,治疗相关不良事件

CC-90010(一种可逆性口服 BET 抑制剂)针对晚期实体瘤和复发/难治性非霍奇金淋巴瘤患者的 I 期研究
背景:溴结构域和额外末端 (BET) 蛋白是表观遗传读取器,可调节参与致癌作用的基因的表达。CC-90010 是一种新型口服可逆小分子 BET 抑制剂。患者和方法:CC-90010-ST-001 (NCT03220347; 2015-004371-79) 是 CC-90010 在晚期或不可切除实体瘤和复发/难治性 (R/R) 非霍奇金淋巴瘤 (NHL) 患者中的 I 期剂量递增和扩展研究。我们报告了剂量递增阶段的结果,该研究探索了 11 个剂量水平和 4 个给药方案,其中两个每周一次(2 天服药/5 天停药;3 天服药/4 天停药),一个每两周一次(3 天服药/11 天停药),以及一个每月一次(4 天服药/24 天停药)。主要目标是确定安全性、最大耐受剂量 (MTD) 和/或推荐的 II 期剂量 (RP2D) 和时间表。次要目标是评估早期抗肿瘤活性的信号、药代动力学和药效学。结果:本研究招募了 69 名患者,其中 67 名患有实体瘤,2 名患有弥漫大 B 细胞淋巴瘤 (DLBCL)。中位年龄为 57 岁(范围,21 至 80 岁),先前治疗方案的中位数为 4 种(范围,1 至 9)。治疗相关不良事件 (TRAE) 大多轻微且可控;超过两名患者报告的 3/4 级 TRAE 是血小板减少症 (13%)、贫血和疲劳(各 4%)。六名患者出现剂量限制性毒性。MTD 分别为 15 毫克(服药 2 天/停药 5 天)、30 毫克(服药 3 天/停药 11 天)和 45 毫克(服药 4 天/停药 24 天)。选定的 RP2D 和扩展方案为 45 毫克(4 天服药/24 天停药)。截至 2019 年 10 月 8 日,1 名 2 级星形细胞瘤患者获得完全缓解,1 名子宫内膜癌患者获得部分缓解,6 名患者病情稳定长达 11 个月。结论:CC-90010 耐受性良好,单药治疗对接受过大量治疗的晚期实体瘤患者有效。关键词:BET 抑制剂、CC-90010、非霍奇金淋巴瘤、实体瘤
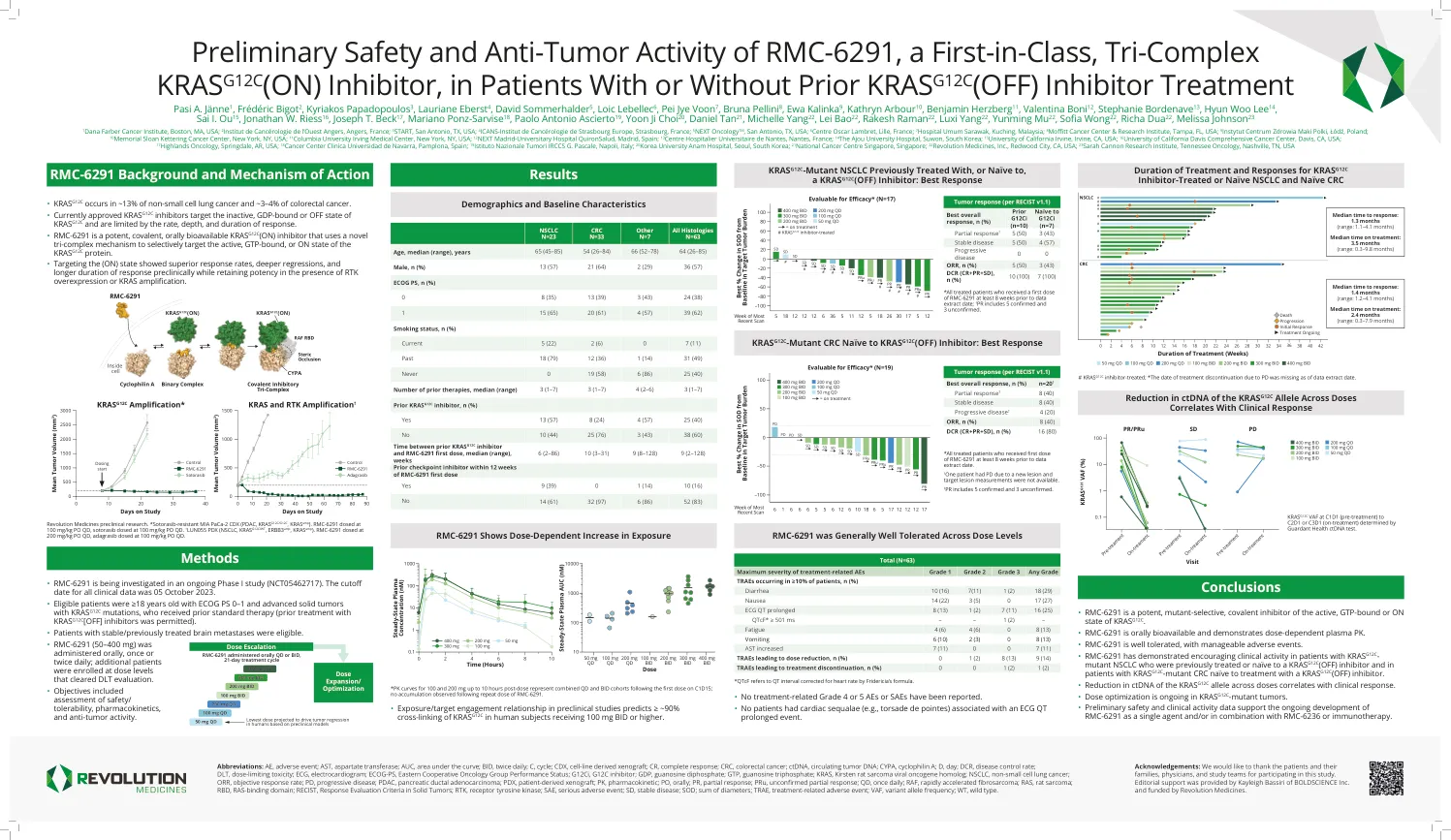
RMC-6291的初步安全性和抗肿瘤活动,A ...
缩写:AE,不利事件; AST,天冬氨酸转移酶; AUC,曲线下方的区域;出价,每天两次; C,循环; CDX,细胞系衍生的异种移植物; CR,完全响应; CRC,结直肠癌; ctDNA,循环肿瘤DNA; CYPA,环磷脂A; D,白天; DCR,疾病控制率; DLT,剂量限制毒性;心电图,心电图; ECOG-PS,东部合作肿瘤学组绩效状况; G12CI,G12C抑制剂; GDP,鸟苷二磷酸盐; GTP,三磷酸鸟嘌呤; Kras,Kirsten大鼠肉瘤病毒癌基因同源物; NSCLC,非小细胞肺癌; ORR,客观响应率; PD,进行性疾病; PDAC,胰腺导管腺癌; PDX,患者衍生的异种移植物; PK,药代动力学; PO,口头; PR,部分反应; PRU,未经证实的部分反应; QD,每天一次; RAF,快速加速的纤维肉瘤;拉斯,老鼠肉瘤; RBD,RAS结合域;恢复,实体瘤的反应评估标准; RTK,受体酪氨酸激酶; SAE,严重的不利事件; SD,稳定疾病;草皮;直径的总和; TRAE,与治疗相关的不良事件; VAF,变体等位基因频率; wt,野生型。

JSKN003 是一种针对 HER2 的抗体-药物偶联物,...
5 (10%) 名患者发生药物相关不良事件,最常见的为腹泻(2.0%)和贫血(2.0%)。2 (4.0%) 名患者发生严重药物相关事件。2 (4.0%) 名患者因药物相关不良事件减少剂量,1 (2.0%) 名患者停药。3 名患者发生药物相关 ILD / 肺炎事件,其中仅 1 名为≥3 级事件。无 TRAE 导致死亡。 在 44 名可评估疗效的患者中,ORR 为 56.8%(95%CI:41.0, 71.7),88.6% (39/44) 的患者肿瘤缩小。当地确诊 HER2 IHC 0 和 HER2 表达(IHC 1+、2+ 和 3+)患者的 ORR 分别为 75%(95% CI:34.9, 96.8)和 52.8 %(95% CI:35.5, 69.6)。中央确诊 HER2 IHC 0 和 HER2 表达患者的 ORR 分别为 52.9%(95% CI:27.8, 77.0)和 68.8%(95% CI:41.3, 89.0)。 33 名接受过贝伐单抗治疗的患者 ORR 为 54.5% (95% CI: 36.4, 71.9),26 名接受过 PARPi 治疗的患者 ORR 为 46.2% (95% CI: 26.6, 66.6)。疗效数据如表 3 所示。 中位随访时间为 2.8 个月,因此中位 PFS 尚未成熟,6 个月 PFS 率为 44.7%。

tivozanib在经过大量预处理的晚期透明细胞肾细胞癌
摘要背景:基于Tivo-3试验,Tivozanib已被批准为晚期肾细胞癌的第三线或后来的疗法,该试验是在免疫检查点疗法(ICT),Cabozantinib和Lenvatinib/everolimus/Everolimus之前进行的,该试验是在当前的顺序治疗paradigm中纳入了Advanced Cliel Cell Rcc(CCRCCC)。方法:我们对6/2021-7/2023中的MD Anderson癌症中心治疗的晚期CCRCC患者进行了回顾性研究。一名盲放射科医生评估了RECIST V1.1的肿瘤反应。我们评估了总体反应率(ORR),临床益处率(CBR)[所有获得放射学反应或稳定疾病(SD)(SD)的患者的百分比≥6个月],无进展生存率(PFS),整体存活率(OS)和安全性。结果:在30名分析患者中,23%的性能状态≥2; 47%的人患有国际转移性RCC数据库财团(IMDC)贫困疾病。先前疗法的中位数为4(范围1-8)。所有患者均接受过先前的ICT,87%的Cabozantinib和60%Lenva tinib±Everolimus。在26名可评估患者中,有2名患者已确认部分反应(ORR 7.7%); 5例患者的SD≥6个月(CBR 23.3%)。中位PFS为3.8个月(范围0.7-13.9);中位OS为14.1个月(范围0.3-28.5)。15例患者(50%)患有≥1例与治疗相关的不良事件(TRAE)。有6级≥3级TRAES [高血压,充血性心力衰竭(3),粘膜炎和GI每锻(5级)]。traes与先前发表的报告一致。结论:在经过大量预处理的患者中,Tivozanib在接受ICT,Cabozantinib和Lenvatinib±依依他的少数患者中产生了适度的临床益处。关键词:肾细胞癌; tivozanib;酪氨酸激酶抑制剂; VEGF封锁;测序。

重组人源化抗HER2的I期研究...
摘要 目的 RC48 含有通过可裂解接头与 MMAE 偶联的新型人源化抗 HER2 抗体 hertuzumab。启动了一项 I 期研究,以评估 RC48 在 HER2 过表达局部晚期或转移性实体癌(特别是胃癌)患者中的毒性、MTD、PK 和抗肿瘤活性。患者和方法这是一项由两部分组成的 I 期研究。连续的患者组接受了递增剂量的 RC48(0.1 mg/kg、0.5 mg/kg、1.0 mg/kg、2.0 mg/kg、2.5 mg/kg 和 3.0 mg/kg)。剂量扩展以 2.0 mg/kg Q2W 的剂量进行。疗效和安全性组包括所有接受至少一剂 RC48 的患者。结果 共入组 57 例患者,由于 3.0 mg/kg 组终止,无法获得 MTD;2.5 mg/kg Q2W 被宣布为 RP2D。RC48 耐受性良好,最常见的 3 级或更严重的 TRAE 包括中性粒细胞减少症(19.3%)、白细胞减少症(17.5%)、感觉减退(14.0%)和结合胆红素升高(8.8%)。整个研究期间发生了 4 例死亡,其中 3 例被认为与 RC48 有关。总体而言,ORR 和 DCR 分别为 21.0% (12/57) 和 49.1% (28/57)。值得注意的是,HER2 IHC2+/FISH- 患者的反应与 IHC2+/FISH+ 和 IHC3+ 患者的反应相似,ORR 分别为 35.7% (5/14)、20% (2/10) 和 13.6% (3/22)。在接受过 HER2 靶向药物治疗的患者中,RC48 也表现出良好的疗效,ORR 为 15.0% (3/20),DCR 为 45.0% (9/20)。结论 RC48 耐受性良好,在 HER2 阳性实体瘤(包括 HER2 IHC 2+/FISH- 状态的胃癌)中表现出良好的抗肿瘤活性。临床试验信息 NCT02881190。

病例报告:晚期十二指肠腺癌化疗联合靶向治疗及放疗后完全缓解一例
十二指肠腺癌(DA)是极为罕见且侵袭性极强的消化系统恶性肿瘤。由于临床表现缺乏特异性,容易误诊、漏诊,治疗方面也缺乏特异性的共识和推荐,因此常将其合并胃癌、结直肠癌。现报告1例晚期DA患者,接受放化疗联合靶向治疗后获完全缓解(CR)。患者2020年10月根治术后病理诊断为DA,受新冠肺炎疫情影响未能按时接受辅助化疗,术后6个月患者发现多发淋巴结肝脏及腹部转移。考虑病情进展,给予XELOX方案(奥沙利铂+卡培他滨)化疗1周期,1周期治疗后肿瘤标志物持续升高;癌胚抗原(CEA)5.03ng/ml(0~5ng/ml),糖类抗原19-9(CA19-9)747.30U/ml(0~37U/ml)。患者还出现了无法耐受的卡培他滨治疗相关不良事件(TRAE),即手足综合征。针对以上原因,患者在第2周期将卡培他滨换为S-1,化疗方案变为SOX(奥沙利铂+S-1);在SOX方案中同时加入贝伐单抗注射液,继续以SOX加贝伐单抗的方案定期治疗7个周期。整个治疗期间肝转移灶呈持续缩小趋势;肿瘤标志物亦呈下降趋势。最终患者在第7个周期获得完全缓解(CR)。完成化疗后,对患者腹腔内存在的耐药转移淋巴结进行放疗,共计10次。但患者在放疗过程中出现严重的骨髓抑制和阻塞性黄疸,最终未能完成放疗计划。目前患者继续使用贝伐单抗和S-1维持治疗,复查未见复发或转移。本例晚期DA,

标签,A ator-1017(Evunzekibart)的第一阶段研究,4- ...
抽象背景ATOR-1017(evunzekibart)是一种靶向共刺激受体4-1BB的人类激动剂免疫球蛋白G4抗体(CD137)。ATOR-1017在肿瘤环境中激活T细胞和天然杀伤细胞,从而导致免疫介导的肿瘤细胞死亡。在这是一个人类,多中心,I期研究的方法中,ATOR-1017每21天静脉内服用ATOR-1017作为单一疗法,以对患有多种护理标准治疗的晚期,无法切除的实体瘤患者进行单一疗法。该研究使用单个患者队列进行快速剂量升级高达40 mg;此后,经过改进的3+3设计最大900毫克。升级剂量,直到疾病进展,不可接受的毒性或戒断同意。研究的主要目标包括通过评估不良事件和限制剂量毒性(DLTS)来确定最大耐受剂量(MTD)。次要目标包括确定药代动力学,免疫原性和使用CT扫描评估的临床疗效,使用实体瘤中的免疫反应评估标准进行了评估。探索性目标包括对免疫系统生物标志物的药效学(PD)评估。筛查的27例患者的结果,25例接受了ATOR-1017的治疗。研究的中位时间为13.1周(范围4.3-92.3)。未达到ATOR-1017的MTD。在25名患者中有13例(52%)报告了与治疗相关的不良事件(TRAES);最常见的(≥10%)是疲劳(n = 4(16.0%))和中性粒细胞减少症(n = 3(12.0%)患者)。没有因Traes而停止的患者,也没有观察到DLT。五名患者经历了严重的(3级)TRAE;中性粒细胞减少症(n = 2),热中性粒细胞减少症(n = 1),胸痛(n = 1),肝酶增加(n = 1),白细胞减少症和血小板减少症(n = 1)。药代动力学数据显示出近似的剂量 - 偏移动力学。PD生物标志物(包括可溶的4-1BB)的剂量依赖性增加表示靶向介导的生物学活性。最佳反应是25例患者中有13例(52%)的稳定疾病,在6例患者中维持6个月或更长时间(24%)。ATOR-1017的结论治疗在所有剂量水平上都是安全且耐受性的,并且表现出生物学活性。此外,在经过大量预处理的人群中,几乎三分之一的患者经历了持久的稳定疾病。令人鼓舞的安全性和初步疗效数据保证

证券交易所公告
肿瘤的生长和进展并提高耐受性,弥补当前治疗方法的差距 • 此次收购扩充了 GSK 在胃肠道 (GI) 癌症领域的产品组合 • GSK 将支付高达 11.5 亿美元 GSK plc (LSE/NYSE: GSK) 和 IDRx, Inc. (IDRx) 今天宣布,他们已达成协议,GSK 将收购 IDRx,这是一家位于波士顿的临床阶段生物制药公司,致力于开发用于治疗 GIST 的精准疗法。根据协议,GSK 将预付 10 亿美元,并有可能在成功获得监管批准后额外支付 1.5 亿美元的里程碑付款。此次收购包括先导分子 IDRX-42,这是一种高度选择性的 KIT TKI,正在开发作为治疗 GIST 的一线和二线疗法。 GIST 通常出现在胃肠道中,80% 的病例是由 KIT 基因突变引起的,这种突变会导致肿瘤细胞生长、增殖和存活(原发性或激活突变)。1 90% 的一线治疗患者会出现新的 KIT 突变(继发性或耐药性突变),这通常会导致复发,治疗选择有限。2 目前,尚无获批的 TKI 可以抑制 KIT 中临床相关的所有原发性和继发性突变。IDRX-42 已证明对所有关键的原发性和继发性 KIT 突变均有效,旨在改善 GIST 患者的预后。这种广泛的突变覆盖范围,加上可以提高耐受性的高选择性,为同类最佳产品提供了潜力。GSK 首席商务官 Luke Miels 表示:“IDRX-42 补充了我们不断增长的胃肠道癌症产品组合。此次收购符合我们收购资产的方法,这些资产针对的是经过验证的目标,并且尽管已有获批产品,但仍存在明显未满足的医疗需求。” GSK 首席科学官 Tony Wood 表示:“IDRX-42 的早期数据及其独特的能力让我们感到兴奋,该能力可以针对 GIST 中存在的所有临床相关 KIT 突变,而这是当前标准治疗的一个重大差距。我们期待在 2025 年加速其开发,以重新定义治疗方法。”StrateGIST 1 的最新临床数据是正在进行的 IDRX-42 对晚期 GIST 患者的 I/Ib 期试验,在结缔组织肿瘤学会 (CTOS) 2024 年年会上以口头报告的形式进行了介绍。这些数据显示,IDRX-42 对晚期 GIST 患者具有良好的抗肿瘤活性,且安全性可控。在二线或更大 GIST 患者中以及所有 KIT 突变亚组中,根据改良 RECIST v1.1 标准,总疗效可评估人群的客观缓解率 (ORR) 为 29% (n=87),包括 1 例完全缓解 (CR) 和 24 例部分缓解 (PR)。在接受过一线治疗的患者中,ORR 为 53% (n=15),其中包括 1 例 CR 和 7 例 PR。3 在所有患者中,在数据截断时,两个 PR 正在等待确认,随后均得到确认。StrateGIST 1 的新耐久性数据也令人满意。IDRX-42 通常耐受性良好,在推荐的 Ib 期剂量下,治疗相关不良事件 (TRAE) 主要为低级别。4