XiaoMi-AI文件搜索系统
World File Search Systemfda

行业和 FDA 员工指南草案 - IEEE 802
无线功能的性能 正确及时地传输医疗数据和信息对于有线和无线医疗设备和系统的安全性和有效性至关重要。但是,符合 IEC 60601- 1-2:2001“医用电气设备 - 第 1-2 部分:安全通用要求 - 并列标准:电磁兼容性 - 要求和测试”的医用电气设备目前不受医疗设备 RF 无线接收器或发射器运行的 RF 频率“排除带”(通带)中的电磁抗扰度规定的限制。这意味着 IEC 60601-1-2:2001 目前不足以评估无线链路在带内 EMD 存在的情况下是否正常运行。因此,FDA 建议您在上市前提交和标签中描述无线技术和 RF 规格(例如,RF 频率和调制)、执行的测试以及您的结果,以证明无线功能将在预期使用环境中安全有效地运行。
ORA微生物学家/生物学家GS-401/403-7/12 FDA的粮食生病爆发的追溯信息 临床评论者:医学博士Patricia Beaston博士 药物开发VICH GL61有关... 的指南 行业指南#116(Vich GL23(R2) breyanzi-包装插入 2024年5月28日564a到期延长信 - jynneos NDP 2024课程Vitae Alik Widge 人类处方药和生物产品标签草案指南中的QTC信息 患者信息 - capvaxive 无标题的字母Krazati
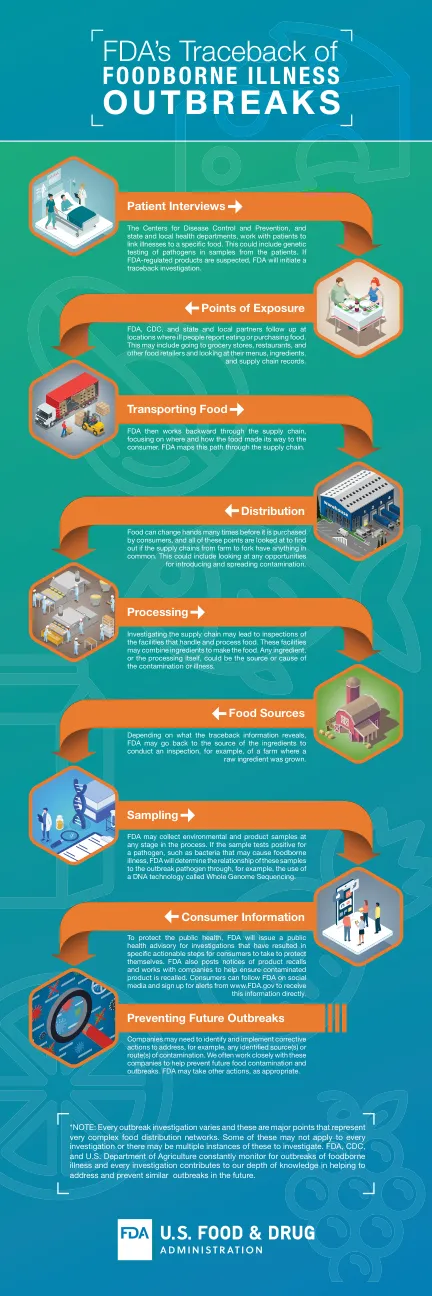
*注意:每次爆发调查都会有所不同,这些是代表非常复杂的食品分配网络的主要点。其中一些可能不适用于每项调查,或者可能有多个实例进行调查。FDA,CDC和美国农业部不断监测食源性疾病的爆发,每项调查都有助于我们在帮助解决和防止将来的类似爆发方面的知识深度。

FDA 的 2023 年重点及未来 Andi Fristedt ...
此后,其他用户付费计划也相继出台,首先是 2002 年的《医疗器械用户付费修正案》(MDUFA),旨在提高医疗器械监管流程的可预测性和透明度,激励创新,让更多产品更快地上市。2012 年颁布的《仿制药用户付费修正案》(GDUFA)旨在帮助缓解由于仿制药申请数量和生产仿制药的外国设施数量增长而导致的上市申请积压问题。同年颁布的《生物仿制药用户付费法案》(BsUFA),授权 FDA 收取费用,以加快生物仿制药申请的审查过程,包括上市后安全活动。用户付费模式还被用来帮助 FDA 审查新型和仿制动物药物的提交内容。这两个用户付费计划都必须在今年重新授权。顺便说一句,对某些烟草产品的国内制造商和进口商征收的使用费将用于资助 FDA 的烟草监管活动,以防止人们开始使用烟草产品,鼓励烟草使用者戒烟并减少吸烟造成的危害。

Optimus 项目 – FDA 的“新”剂量优化和……
• 制定剂量探索和剂量优化策略,利用非临床和临床数据进行剂量选择,包括在试验中对一系列剂量进行随机评估。此类策略的重点是尽可能在开发计划中尽早、尽可能高效地进行这些研究,为患者带来有希望的新疗法。

基于 FDA 不良事件报告的真实世界研究
胃肠穿孔 (GIP) 是一种严重且相对少见的不良事件 (AE),可能会致命。由于药物相关的 GIP 后果严重,因此引起了临床的关注。使用非甾体抗炎药、抗凝剂、皮质类固醇、某些抗肿瘤药物和一些其他药物 ( 1 – 4 ) 都可能导致 GIP。制药行业的发展和创新催生了许多新的有效的抗癌治疗方法,如小分子靶向药物和单克隆抗体。抗体-药物偶联物、程序性细胞死亡蛋白 1 (PD-1)/程序性死亡配体 1 (PD-L1)、嵌合抗原受体 T 细胞 (CRA-T) 等正在迅速崛起,成为肿瘤患者的福音 ( 5 , 6 )。新型抗肿瘤药物引起的胃肠道不良反应已经常被提及,但有关这些新型抗肿瘤药物引起的GIP不良反应却鲜有报道。FDA增加了一个黑框,建议对GIP患者永久停用贝伐单抗(7)。血管内皮生长因子(VEGF)及其受体(VEGFR)的抑制剂与GIP可能相关(8,9)。随着越来越多的新型抗肿瘤药物问世,系统地研究新型抗肿瘤药物与GIP的关系具有重要的临床实际意义。遗憾的是,尚无大规模、全面的研究证实这种关联。美国FDA建立的庞大的不良反应报告数据库为处方药临床应用的安全性提供了丰富的真实世界数据(10)。最重要的是,这个数据库是免费的,向公众开放(11)。数据挖掘是公认的早期发现药物安全信号的良好方法,可用于预测和了解药物的安全性(12)。近年来,基于数据挖掘方法与FDA不良事件报告系统(FAERS)数据库相结合的药物警戒研究日益盛行。本研究利用FAERS数据库,通过计算不良事件信号提供参考依据,以期更好地了解GIP与新型抗肿瘤药物之间的关联,从而确保临床安全使用。


FDA对食品安全和食源性疾病暴发的监督
FDA通常将其食品安全法规基于风险评估框架以控制或防止危害,优先考虑那些污染食物的风险或可能性很高的人。1969年,FDA建立了当前的良好制造实践(CGMP,21 C.F.R. 第110部分)确保食品制造商维护清洁设施并确保食品安全。 FDA此后通过添加预防,监视和记录保存要求来更新CGMP法规。 由于病原体的生长状况和消费习惯,某些食物比其他食物比其他食物更常见于食源性疾病。 FDA发布了特定的法规,以解决低酸罐头食品,酸化食品和贝壳鸡蛋等产品的独特安全问题,重点是某些病原体。 其他特定食品特定的法规,例如果汁和海鲜的法规,基于分析和控制关键控制点(HACCP)特定危害的系统。 在FSMA制定后,FDA发布了其他法规(例如,对人类食品规则的预防控制;制定安全规则;和食品可食用性规则),以减少食品污染,防止食物污染,保留必要的记录,以监控有效性的控制方法,并从市场上删除潜在污染的食品。1969年,FDA建立了当前的良好制造实践(CGMP,21 C.F.R.第110部分)确保食品制造商维护清洁设施并确保食品安全。FDA此后通过添加预防,监视和记录保存要求来更新CGMP法规。由于病原体的生长状况和消费习惯,某些食物比其他食物比其他食物更常见于食源性疾病。FDA发布了特定的法规,以解决低酸罐头食品,酸化食品和贝壳鸡蛋等产品的独特安全问题,重点是某些病原体。其他特定食品特定的法规,例如果汁和海鲜的法规,基于分析和控制关键控制点(HACCP)特定危害的系统。在FSMA制定后,FDA发布了其他法规(例如,对人类食品规则的预防控制;制定安全规则;和食品可食用性规则),以减少食品污染,防止食物污染,保留必要的记录,以监控有效性的控制方法,并从市场上删除潜在污染的食品。
FDA 批准的第一个也是唯一一个可减少蛋白尿的治疗方法......
TARPEYO®(布地奈德)缓释胶囊是一种皮质类固醇,用于降低患有原发性免疫球蛋白 A 肾病 (IgAN) 且有快速病情进展风险的成人患者的蛋白尿,通常尿蛋白与肌酐比 (UPCR) ≥1.5 g/g。该适应症根据蛋白尿减少而获得加速批准。尚未确定 TARPEYO 是否能减缓 IgAN 患者的肾功能衰退。该适应症的继续批准可能取决于确认性临床试验中临床益处的验证和描述。重要安全信息禁忌症:对布地奈德或 TARPEYO 的任何成分过敏的患者禁用 TARPEYO。使用其他布地奈德制剂时曾发生过严重的过敏反应,包括过敏反应。请参阅整个说明书和随附的完整处方信息中的其他重要安全信息。

2011 年至 2019 年 FDA 批准的靶向药物
手性药物通常含有手性中心,以单一对映体或外消旋体的形式存在,与非手性药物相比,其在安全性和有效性方面具有显著优势,且立体选择性高。在这些药物中,手性不仅对溶解度和药代动力学特性有影响,而且对其靶标有特定的机制特征。我们注意到,近十年来,具有独特手性的小分子已成为FDA批准的抗肿瘤药物的新型组分,自批准以来,这些药物不断被探索以用于新适应症、新作用机制和新组合。本文总结了2011年至2019年FDA批准的22个手性小分子靶向抗肿瘤药物的最新研究进展,强调了它们的应用潜力和优势。我们相信这些最新成果可以为优化药物疗效、扩大临床应用、克服耐药性和提高未来手性靶向药物临床给药的安全性提供理论基础并激发研究兴趣。

稀有疾病药物开发的新终点
此外,FDA与明尼苏达大学公共卫生学院签订了签约,以更好地告知爆发与爆发有关的工作。由此产生的独立报告1是基于对25多名FDA高级官员以及美国农业部(USDA)食品安全检验局(FSIS)和疾病控制与预防中心(CDC),州卫生卫生官员以及行业和消费者消费者食品爆发专家的访谈。它提供了对FDA的结构和功能能力的客观评估,以支持,参与或领导多国粮食疾病爆发调查活动。该报告包括一系列建议,在FDA的发展中也发挥了关键作用。