XiaoMi-AI文件搜索系统
World File Search Systemphosphorylation

通过设计为抑制剂的小分子激活 Gcn2
综合应激反应 (ISR) 是细胞保护自己免受环境应激的重要机制。ISR 的核心是一组监测应激条件的相关蛋白激酶,例如 Gcn2 (EIF2AK4) 可识别营养限制,诱导真核翻译起始因子 2 (eIF2) 的磷酸化。Gcn2 磷酸化 eIF2 可降低大部分蛋白质合成,节省能量和营养,同时优先翻译应激适应基因转录本,例如编码 Atf4 转录调节因子的转录本。虽然 Gcn2 对细胞保护免受营养应激至关重要,并且其在人类中的消耗会导致肺部疾病,但 Gcn2 还可能导致癌症进展并在慢性应激期间促进神经系统疾病。因此,已经开发出特定的 ATP 竞争性 Gcn2 蛋白激酶抑制剂。在本研究中,我们报告了一种这样的 Gcn2 抑制剂 Gcn2iB 可以激活 Gcn2,并且我们探究了这种激活发生的机制。低浓度的 Gcn2iB 会增加 eIF2 的 Gcn2 磷酸化并增强 Atf4 的表达和活性。重要的是,Gcn2iB 可以激活缺乏功能性调节域或具有某些激酶域替换的 Gcn2 突变体,这些突变体源自缺乏 Gcn2 的人类患者。其他 ATP 竞争性抑制剂也可以激活 Gcn2,尽管它们的激活机制有所不同。这些结果为 eIF2 激酶抑制剂在治疗应用中的药效学提供了警告。旨在直接激活 Gcn2 的激酶抑制剂化合物,甚至是功能丧失的变体,可以提供缓解 Gcn2 和 ISR 其他调节剂缺陷的工具。

1介导减数分裂双链破裂活动
在第一个减数分裂细胞分裂中摘要,大多数生物体的染色体的适当分离取决于chiasmata,这是源自spo11核酸酶催化的编程双链断裂(DSB)的同源染色体之间的连续性交换。由于DSB会导致生殖细胞无法弥补的损害,而缺乏DSB的染色体也缺乏Chiasmata,因此必须仔细调节DSB的数量既不会太高也不太低。在这里,我们表明,在秀丽隐杆线虫中,减数分裂DSB水平受DSB-1的磷酸调节控制,DSB-1是PP4 PPH-4.1磷酸酶和ATR ATL-1 Kinase的相对活性,DSB-1(酵母SPO11辅助剂REC114)的同源物。PPH-4.1突变体中DSB-1磷酸化的增加与DSB形成的减少相关,而DSB-1磷酸化的预防大大增加了PPH-4.1突变体和野生型背景中的减数分裂DSB的数量。秀丽隐杆线虫及其近亲还具有DSB-1的差异旁系同源物,称为DSB-2,而DSB-2的丢失却可以减少年龄增加的卵母细胞中的DSB形成。我们表明,DSB-1的哲学和灭活形式的比例随着年龄的增长和DSB-2的流失而增加,而不可磷酸化的DSB-1则挽救了DSB-2突变体中DSB的年龄依赖性降低。这些结果表明,DSB-2部分进化以补偿DSB-1通过磷酸化的失活,以维持老年动物的DSB水平。我们的工作表明,PP4 PPH-4.1,ATR ATL-1和DSB-2与DSB-1协同作用,以在整个生殖寿命中促进最佳DSB水平。

JAK抑制剂的新兴世界
Janus激酶(JAK)蛋白是酪氨酸激酶蛋白,与信号换能器和转录(Stat)蛋白的激活剂一起形成JAK STAT途径。Jak家族有四个成员:JAK1,JAK2,JAK3和TYK2; STAT家族有7个成员。JAK蛋白位于跨膜蛋白的细胞内部分作为同型或异二聚体。当信号分子附着在跨膜蛋白上时,它会随着JAK分子的磷酸化而经历结构变化。这些JAK分子然后形成了统计蛋白的对接位点。stat蛋白然后经历磷酸化,转移到细胞核并调节基因转录。1 JAK途径失调与各种自身免疫性疾病有关。jak抑制剂(Jaki)是小分子,由于其在免疫发作中的选择性作用,其作用很广。这些分子可作为口服或局部药物提供,增加了它们的可接受性和便利性。jaki已获得FDA的批准,用于治疗特应性皮炎(口服abrocitinib,口服upadacitinib和局部ruxolitinib),脱发

[DZNE-2024-00092.pdf
摘要:神经退行性疾病,例如帕金森氏病,阿尔茨海默氏病和亨特顿病,都以神经元和神经元功能障碍的进行性丧失鉴定和特征,导致认知和运动障碍。最近的研究表明,PTM的重要性,例如磷酸化,乙酰化,甲基化,泛素化,Sumoylation,硝化,硝化,截断,O-Glcnacylation和羟基化和羟基化,在NeuroDegeneration灾难的进展中。PTM可以改变蛋白质的结构和功能,从而影响蛋白质稳定性,定位,相互作用和酶活性。异常的PTM会导致蛋白质错误折叠和聚集,降解和清除,并最终导致神经元功能障碍和死亡。本综述的主要目的是概述与神经变性有关的PTM,其潜在机制,分离PTM的方法以及这些疾病的潜在治疗靶标。本文讨论的PTM包括tau磷酸化,α-突触核蛋白和狩猎蛋白泛素,组蛋白乙酰化和甲基化以及RNA修饰。了解PTM在神经退行性疾病中的作用可能为这些毁灭性疾病提供新的治疗策略。

糖尿病心肌病中代谢失调的分子机制
糖尿病性心肌病(DCM)是糖尿病最严重的并发症之一,已被认为是一种心脏代谢疾病。在常规条件下,心跳加速所需的大多数ATP产生(> 95%)来自脂肪酸(FAS)和葡萄糖的线粒体氧化磷酸化,其余部分来自各种来源,包括果糖,包括果糖,乳酸,乳酸酮(Ketone Body)(Ketone Body(KB)和分支链型氨基酸(BCAA)(BCAA)。在动物模型和糖尿病患者的糖尿病心脏中观察到了葡萄糖和乳酸的摄入量增加,并降低了葡萄糖和乳酸的利用率。此外,聚元途径被激活,果糖代谢得到增强。将酮用作人类糖尿病心脏中的能源也显着增加。此外,在糖尿病小鼠和患者的心脏中观察到BCAA水平升高和BCAA代谢受损。糖尿病心脏中能量底物偏好的转移会导致氧气消耗量增加和氧化磷酸化受损,从而导致糖尿病性心肌病。但是,

人肺肿瘤衍生细胞模型中蛋白激酶 AKT 和 ERK1/2 之间的串扰
毫无疑问,细胞信号操控是抗癌治疗的关键策略。此外,细胞状态决定药物反应。因此,建立细胞状态和治疗敏感性之间的关系对于癌症疗法的发展至关重要。在个性化医疗时代,使用患者来源的离体细胞模型是将关键研究成果转化为临床应用的一种有前途的方法。在这里,我们专注于细胞对抗癌治疗耐药性的非致癌基因依赖性。使用一组具有各种干细胞和 EMT 相关标志物、不同程度的 ERK1/2 和 AKT 磷酸化以及对抗癌治疗反应的患者肺肿瘤衍生细胞系研究了对 MEK/ERK 和 PI3K/AKT 通路抑制剂(关键细胞功能调节剂)的反应信号相关机制。研究激酶之间的相互作用是我们研究的目标。尽管 MEK/ERK 和 PI3K/AKT 相互作用被认为是细胞系特异性的,其中致癌突变起着决定性作用,但我们证明了所有研究的细胞系中 MEK/ERK 和 PI3K/AKT 信号通路之间存在负反馈回路,无论基因型和表型差异如何。我们的研究表明,各种不同的 ERK 信号抑制剂(selumetinib、trametinib 和 SCH772984)可增加 AKT 磷酸化,相反,AKT 抑制剂(capivasertib、idelalisib 和 AKT 抑制剂 VIII)可增加对照细胞和顺铂治疗细胞中的 ERK 磷酸化。然而,激酶之间的相互作用取决于细胞状态。 ERK 和 AKT 之间的反馈被局部粘连激酶抑制剂 PF573228 减弱,并且在悬浮生长的细胞中也是如此,这表明细胞外接触在调节激酶之间的串扰方面可能发挥着作用。此外,研究表明,MEK/ERK 和 PI3K/AKT 信号通路之间的相互作用可能取决于化疗刺激的强度。该研究强调了抗癌治疗期间细胞的空间位置和治疗强度的重要性。

微管活性调节激酶 2 增强宿主……
先天免疫系统对于抵御病原体入侵、有效控制感染以及触发适应性免疫反应以消除传染源至关重要。本研究揭示了微管亲和力调节激酶 2 (MARK2) 作为广谱抗病毒免疫调节剂的关键作用,具体通过其与鸟嘌呤核苷酸交换因子 H1 (GEF- H1) 的相互作用以及与 TANK 结合激酶 1 (TBK1) 的结合。至关重要的是,MARK2 的抗病毒功效取决于其激酶活性,特别是其在丝氨酸 645 位点磷酸化 GEF-H1 的能力。该磷酸化事件是激活 TBK1 的关键触发因素,从而导致诱导 I 型干扰素 (IFN-I) 和干扰素刺激基因 (ISG)。我们的结果表明,GEF-H1 是一种 ISG,并由 MARK2 促进。这些发现不仅证实了 MARK2 是 GEF-H1 的激酶,还揭示了 MARK2 增强宿主抗病毒防御的一种以前未被认识的机制。通过对 GEF-H1 进行策略性磷酸化来增强 IFN-I 信号,MARK2 显著增强了抗病毒免疫反应,为细胞防御机制的分子协调提供了新的见解。

使用基于DNA的验光工具对受体信号传导动力学的非核控制
生长因子(GFS)是多肽配体,这些配体调节各种细胞活性,例如增殖,迁移和分化。在细胞膜上GFS与受体酪氨酸激酶(RTK)的结合可诱导RTK的二聚化和随后的磷酸化,并启动细胞内激酶的磷酸化级联反应。1有趣的是,RTK下游激酶的激活动力学在确定细胞功能和命运方面起着重要作用(图1)。2例如,在大鼠PC12细胞中,表皮生长因子(EGF)和神经生长因子(NGF)都通过激活其同源受体激活RAS-RAF-MEKERK途径,但是激酶激活的动力学以及所得的细胞灭绝表现出独特的模式。3 EGF导致ERK的瞬时激活,导致细胞增殖,而NGF导致ERK的持续激活,从而导致细胞分化。迄今为止,已经关注了启动GF信号动力学的机制,从而调节细胞功能。因此,高度寻求能够控制具有精确时间分辨率的RTK活性的方法,以研究受体 -
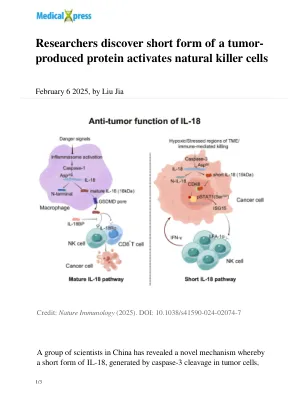
产生的蛋白质激活天然杀伤细胞
在这项研究中,研究人员专注于IL-18。他们发现肿瘤细胞可以通过caspase-3裂解产生新型的IL-18形式,该裂解与传统的成熟IL-18途径无关。与成熟的IL-18不同,这种短形式不会退出细胞,而是进入细胞核,在该细胞核中促进了STAT1和ISG15分泌的磷酸化,从而增强了NK细胞抗肿瘤功能。

CDK5 通过分子伴侣破坏 PD- L1
摘要 背景 在过去的几年中,针对程序性细胞死亡蛋白 1 (PD-1) 及其配体程序性细胞死亡配体 1 (PD-L1) 的肝细胞癌 (HCC) 免疫疗法已经取得了持久的临床效益。然而,仅有一小部分 HCC 患者对单独 PD-1/PD-L1 阻断表现出客观的临床反应。尽管对 PD-L1 翻译后修饰的影响很大,但其在 HCC 免疫疗法耐药性中的意义仍不甚明了。方法 在 HCC 细胞中敲低细胞周期蛋白依赖性激酶 5 (CDK5) 表达,用蛋白质印迹法检测 CDK5 和 PD-L1 蛋白水平。进行免疫共沉淀以评估蛋白质之间的相互作用。构建临床前 HCC 小鼠模型以评估 CDK5 抑制剂单独或与 PD-1 抗体联合使用的效果。使用临床HCC样本阐明CDK5、PD-L1和PD-L1 T290磷酸化在HCC中的临床意义。结果我们发现CDK5缺陷会上调HCC细胞中PD-L1蛋白的表达,并揭示出一种PD-L1被CDK5下调的新型分子机制,即CDK5介导的T290位PD-L1磷酸化促进其与伴侣蛋白热休克同源蛋白70(HSC70)结合并通过伴侣介导的自噬进行降解。值得注意的是,CDK5抑制剂PNU112455A治疗可有效上调肿瘤PD-L1水平,促进对抗PD-1免疫治疗的反应,并延长HCC肿瘤小鼠的生存时间。此外,PD-L1的T290磷酸化状态与HCC的预后相关。结论 靶向CDK5可与PD-1阻断协同抑制HCC生长,可能具有临床益处。本研究揭示了HCC中PD-L1降解的独特调控,为HCC的临床治疗提供了一个有吸引力的治疗靶点、一种潜在的药物和一种新的预后指标。