XiaoMi-AI文件搜索系统
World File Search SystemsgRNA

sgrnas
与其他冠状病毒一样,SARS-COV-2基因表达策略是基于嵌套亚基因组mRNA物种(SGRNA)的合成。这些SGRNA是使用“不连续的转录”机制合成的,该机制依赖于在转录调节序列(TRS)下切换的模板切换。可以产生典型(c- sgrna)和非典型的(NC-SGRNA,较少)亚基因组RNA物种。当前,根据下一代测序(NGS)获得的序列数据研究了SGRNA,生物信息学工具对于它们的识别,表征和定量至关重要。迄今为止,该目标很少有软件,他们的可靠性和适用性需要建立在所有可用的NGS平台上,以建立对这些工具产生的信息的信心。实际上,这些信息可能对阐明病毒表达策略的深入阐明至关重要,尤其是在NC-SGRNA的意义上,以及可能将SGRNA用作感染患者病毒复制活性的潜在标记。
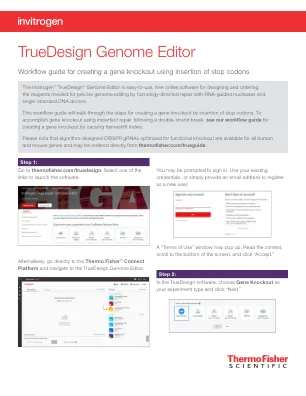
TrueDesign 基因组编辑器
视图将更新以显示“选择 gRNA 选项”。单击适合您需求的 sgRNA 选项 - “预先设计的 sgRNA”(仅限人类和小鼠基因)或“自定义 gRNA 和 TALEN 设计”。如果您对插入终止密码子的区域没有偏好,并且正在人类或小鼠细胞中工作,建议使用“预先设计的 sgRNA”。否则,选择“自定义 gRNA 和 TALEN 设计”。本文档中显示的大多数步骤对于两个工作流程都相似。

AccuTool™ sgRNA-(GFP) 合成 (dRGEN)
DNA 引导的 RNA 引导内切酶 (dRGEN) 是高效、经济且方便的基因组编辑实验工具。AccuTool™ dRGEN 可识别长度为 23 bp 并以两个鸟嘌呤 (GG) 结尾的目标序列。定制 sgRNA 表达质粒可与 Cas9 表达质粒(人类密码子优化,WT/Nickase/Sniper 形式可用)一起使用。质粒可以通过任何标准方法(如脂质转染、纳米颗粒或电穿孔)递送到您感兴趣的细胞中,以实现高效递送。sgRNA-GFP 表达质粒是通过将 GFP 构建体插入现有 sgRNA 质粒中构建的。sgRNA-GFP 表达质粒可让您通过荧光显微镜确认细胞中的活性水平。 AccuTool™ dRGEN 是一种定制设计的 sgRNA,以 sgRNA 表达质粒或 sgRNA-GFP 表达质粒的形式提供。应用

1 LeishGEdit 工具箱的 Bar-seq 策略 2
LeishGEdit 引物设计流程为基因组中每个给定的 ORF 设计了总共六个引物序列,以实现 CRISPR-Cas9 基因编辑,从而允许用大量可用标签标记感兴趣基因的 N 端或 C 端,并删除 ORF 的两个等位基因(图 1B 和 C)。设计了两个 sgRNA 引物,一个靶向目标基因的 5'UTR,一个靶向 3'UTR。sgRNA 引物由 T7 RNAP 启动子序列、20 nt sgRNA 靶序列(用于在感兴趣的位点引入 DSB)和 20 nt 与 CRISPR-Cas9 主链序列的重叠组成,从而允许通过 PCR 生成 sgRNA 模板。需要一个包含整个 sgRNA 主链序列 [20] 的额外通用引物来扩增两个 sgRNA。四种引物专为 pPLOT 和 pT 质粒扩增而设计,可以以不同的组合使用 82 来产生供体 DNA。这些引物包括:上游正向引物 (#1)、83 上游反向引物 (#2)、下游正向引物 (#3) 和下游反向引物 (#4)。可以选择性地设计 84 额外引物,以允许使用从 pPOT 质粒模板扩增的供体 85 构建体进行 CRISPR-Cas9 介导的基因编辑 [21]。供体 DNA 引物包含紧邻 sgRNA 靶序列及其 PAM 位点的 30 nt HF 序列 86,以及与 pT、pPLOT 和 pPOT 质粒兼容的引物结合位点 87。虽然上游正向引物和下游 88 反向引物位置始终根据所选的 sgRNA 而变化,但上游反向引物 89 (#2) 和下游正向引物 (#4) 针对每个基因设计在相同的位置。90

e CIENT CRISPR/CAS9介导的合并-sgrnas ...
结果:在一个实验中靶向多个基因,通过优化的CRISPR/CAS9系统将112个与植物发育相关的基因拆除。我们优化了合并的SGRNA组装方法的关键步骤,该方法通过该方法将116个SGRNA合并为4组(每组由29个SGRNA组成)。每组SGRNA都是在一个PCR反应中编译的,随后经过一轮矢量构建,转化,SGRNA鉴定以及一轮遗传转化。通过遗传转化介导的农杆菌,我们成功地产生了800多个植物。 对于突变体识别,已经使用了下一代测序技术,结果表明所有产生的植物都是阳性的,并且涵盖了所有靶向基因。 有趣的是,在所有转基因植物中,85%通过遗传转化介导的农杆菌,我们成功地产生了800多个植物。对于突变体识别,已经使用了下一代测序技术,结果表明所有产生的植物都是阳性的,并且涵盖了所有靶向基因。有趣的是,在所有转基因植物中,85%

不耐受的不耐受性使有效的微生物单基因组编辑
摘要:CRISPR/CAS9系统最近已成为一种有用的基因特定编辑工具。然而,这种方法偶尔会导致由于不匹配耐受性而导致的DNA靶标和类似的DNA序列消化,这仍然是当前基因组编辑技术的显着缺点。但是,我们的研究确定,即使是靶DNA和5'截断的SGRNA之间的单基碱基不匹配也抑制了靶标识别。这些结果表明,5'截断的SGRNA/CAS9复合物可用于在微生物基因组中进行负选择单基本编辑的靶标。此外,我们证明了5'截断的SGRNA方法可用于简单有效的单基本编辑,因为它可以对DNA靶标的单个碱基进行修改,远离截短SGRNA的5'端。此外,当使用具有膨胀的原始探针邻近基序(PAM; 5'-NG)的工程Cas9核酸酶时,还允许5'截断的SGRNA进行有效的单基础编辑,这可以启用全基因组单基础量表。
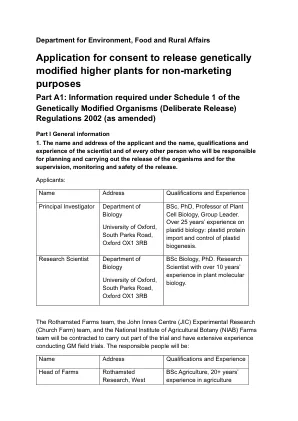
A Part A1-牛津大学申请发行GMO(参考24/R57/01)
引入的构建体是通过CRISPR/CAS9系统诱导基因靶向敲除突变体的质粒载体。根据Smedley等人组装该质粒载体。,2021通过使用金门(GG,类型IIS限制酶)模块化克隆(MOCLO)组件。用于针对每个亚基因组中感兴趣基因的三个同源副本(a,b和d),在三个亚基因组的第一个外显子中选择了两个单个引导RNA(SGRNA)序列(sgrNA)序列(guide_2和gude_3)。如Smedley等人所述,使用WheatCrispr工具用于选择SGRNA。,2021。考虑了预测的目标和脱靶切割效率;预计SGRNA脱离目标得分的值为“ 0”,相当于根据WheatCrispr工具在其他遗传或基因间区域中的“无预测命中”。

CRISPR 转录激活因子的特殊调节......
摘要 细胞转录本编码有关细胞身份和疾病状态的重要信息。响应 RNA 生物标志物激活 CRISPR 有可能以时空精度控制 CRISPR 活性。这将能够将 CRISPR 活性限制在表达目标 RNA 生物标志物的特定细胞类型,同时防止其他细胞中出现不必要的活性。在这里,我们提出了一个简单而具体的平台,用于通过工程化脓性链球菌 Cas9 单向导 RNA (sgRNA) 来调节响应 RNA 检测的 CRISPR 活性。sgRNA 被设计成折叠成复杂的二级结构,在基态下抑制其活性。工程化的 sgRNA 在识别互补 RNA 后被激活,从而使 Cas9 能够发挥其功能。我们的方法使 CRISPR 能够在 HEK293T 细胞和斑马鱼胚胎中响应 RNA 检测而激活。迭代 21 设计优化允许开发用于生成能够检测所选 RNA 序列的 sgRNA 22 的计算工具。机制研究表明,工程 23 sgRNA 在 RNA 检测过程中被切割,并且我们确定了受益于 24 化学修饰的关键位置,以提高工程 sgRNA 在体内的稳定性。我们的传感器为使用 CRISPR 26 激活来响应内源性 RNA 生物标志物开发新的研究和治疗应用开辟了新的机会。 27

ret融合基因阳性肺癌的新分子靶耐药机制...
接下来,使用LCC-190细胞进行CRISPR/CAS9的整个基因组敲除筛选,以鉴定与抗RET抑制剂抗性相关的基因。将LCC-190细胞与大约120,000个SGRNA文库(包含3-6个SGRNA敲除一个基因)一起引入,用RET抑制剂处理了大约9天,然后检查了幸存细胞中包含的SGRNA。结果强烈表明,ERRFI1(MIG6)基因的敲除参与RET抑制剂耐药性(图2)。为了验证这一结果,使用LC2/AD和LCC-190细胞用具有不同序列的其他SGRNA敲除Mig6。结果表明,在Mig6基因敲除细胞中,EGFR途径过度活化,诱导耐药性。 EGF以1 ng/ml的浓度共同治疗,与人类的血液浓度相当

CRISPR 转录激活因子的特定调节......
摘要 细胞转录本编码了有关细胞身份和疾病状态的重要信息。响应 RNA 生物标志物而激活 CRISPR 有可能以时空精度控制 13 CRISPR 活性。这将能够将 CRISPR 活性限制在表达目标 RNA 生物标志物的特定细胞类型,同时防止其他细胞中出现不必要的活性。在这里,我们提出了一个简单而具体的平台,用于通过工程化脓性链球菌 Cas9 单向导 RNA (sgRNA) 来调节响应 RNA 检测的 CRISPR 活性。sgRNA 被设计成折叠成复杂的二级结构,在基态下抑制其活性。识别互补 RNA 后,工程化的 sgRNA 19 被激活,使 Cas9 能够发挥其功能。我们的方法使 CRISPR 20 在 HEK293T 细胞和斑马鱼胚胎中响应 RNA 检测而激活。迭代 21 设计优化允许开发用于生成能够检测所选 RNA 序列的 sgRNA 22 的计算工具。机制研究表明,工程 23 sgRNA 在 RNA 检测过程中被切割,并且我们确定了受益于 24 化学修饰的关键位置,以提高工程 24 sgRNA 在体内的稳定性。我们的传感器为使用 26 CRISPR 激活来响应内源性 RNA 生物标志物开发新的研究和治疗应用开辟了新的机会。 27