XiaoMi-AI文件搜索系统
World File Search SystemsgRNA

通过计算机设计 CRISPR/Cas9 引导 RNA 来敲除红薯 (Ipomoea batatas L.) 中的八氢番茄红素去饱和酶基因
摘要 本研究旨在设计计算机引导RNA(sgRNA),用于CRISPR/Cas9介导的红薯(Ipomoea batatas L.)八氢番茄红素脱氢酶(PDS)基因敲除。IbPDS基因编码区序列长1791个碱基对(bp),相当于572个氨基酸。将IbPDS基因的氨基酸序列与其他邻近植物物种的同源序列进行比较,结果显示,它与Ipomoea triloba和Ipomoea nil的PDS相似性很高,分别为98.60%和97.73%。 CRISPR RGEN Tools 为 IbPDS 基因提供了 113 个结果,筛选出 24 个,并选择了三个 sgRNA 序列用于设计基因编辑载体,它们是 sgRNA 1 (5'-AC- CTCATCAGTCACCCTGTCNGG-3')、sgRNA 2 (5'- CCTCCAGCAGCAGTATTGGTTGGTTTGNGG -3') 和 sgRNA 3 (5'- CTGAACTCTCCTGGTTGGTTGTTNGG -3')。所选 sgRNA 的预测二级结构为靶基因的基因编辑提供了有效的 sgRNA 结构。用于 CRISPR/Cas9 介导的 IbPDS 基因敲除的 PMH-Cas9- 3xsgRNA 载体是使用三个 sgRNA 序列和一个潮霉素抗性标记在计算机上设计的。

SNP-CRISPR:用于 SNP 特异性基因组编辑的网络工具
摘要 CRISPR-Cas9 是一种强大的基因组编辑技术,其中单个向导 RNA (sgRNA) 赋予靶位点特异性以实现 Cas9 介导的基因组编辑。已经基于人类和模型生物的参考基因组开发了大量 sgRNA 设计工具。然而,现有资源并不是最佳的,因为靶向区域内的基因突变或单核苷酸多态性 (SNP) 会干扰向导-靶互补性,从而影响基于 CRISPR 的方法的效率。为了便于识别 (1) 非参考基因组中的 sgRNA、(2) 不同遗传背景下的 sgRNA 或 (3) 针对含 SNP 的等位基因的特定靶向,例如疾病相关突变,我们开发了一个网络工具 SNP-CRISPR ( https://www.fl yrnai.org/tools/snp_crispr/ )。 SNP-CRISPR 可用于根据公共变异数据集或用户识别的变异设计 sgRNA。此外,该工具还计算针对变异和参考的 sgRNA 设计的效率和特异性得分。此外,SNP-CRISPR 提供了上传多个 SNP 的选项,并使用单个 sgRNA 设计同时针对一个或多个附近的碱基变化。鉴于这些功能,SNP-CRISPR 在模型系统中以及用于疾病相关变异校正的 sgRNA 设计中具有广泛的潜在研究应用。
GeneTargeter:在疟原虫寄生虫中的基因组编辑中自动化,恶性疟原虫
丰富度与更好的编辑活动有关[54,55]。均聚物据报道偶尔会降低SGRNA效率[54-56]。 可以用两种算法之一来计算sgrna裂解预期位点的预测概率的目标分数:(1)Doench等人开发的原始规则集2分数。 cas9 sgrnas [57],并以方位角更新(github.com/microsoftresearch/azimuth);或(2)用于与CAS12A SGRNA一起开发的Cindel分数[53]。 最后,可用的靶向活动评分算法包括HSU等人开发的分数。 [58]和Doench等人开发的切割确定(CFD)得分。 [57]。 两者都是基于选择的SGRNA与目标基因组中所有其他可能的SGRNA之间成对比较的分数,并且使用系数矩阵确定成对得分,该系数矩阵在SGRNA中考虑了不匹配位置,以及在CFD得分的情况下确定了不匹配的身份。 因为两个分数的系数矩阵均来自均聚物据报道偶尔会降低SGRNA效率[54-56]。可以用两种算法之一来计算sgrna裂解预期位点的预测概率的目标分数:(1)Doench等人开发的原始规则集2分数。cas9 sgrnas [57],并以方位角更新(github.com/microsoftresearch/azimuth);或(2)用于与CAS12A SGRNA一起开发的Cindel分数[53]。最后,可用的靶向活动评分算法包括HSU等人开发的分数。[58]和Doench等人开发的切割确定(CFD)得分。[57]。两者都是基于选择的SGRNA与目标基因组中所有其他可能的SGRNA之间成对比较的分数,并且使用系数矩阵确定成对得分,该系数矩阵在SGRNA中考虑了不匹配位置,以及在CFD得分的情况下确定了不匹配的身份。因为两个分数的系数矩阵均来自
Monarch®RNA清理套件
使用Monarch RNA清洁套件(NEB#T2040)或竞争对手试剂盒(根据制造商的建议)清理了Engen®SgrNA合成试剂盒(NEB#E3322)的六种不同的SGRNA合成反应(NEB#E3322)(NEB#E3322),并在50μL的50μLNutece-Free Free Water中进行了体重。SGRNA产量是根据由Trinean Dropsense™16进行测量的结果A260计算的。君主RNA清理套件产生的SGRNA产生与其他市售RNA清理套件一致的。清理后,通过LC-MS测量残留核苷酸(NTP),并报告为面积为NTPS百分比(RATP+RCTP+RGTP+RUTP)/SGRNA百分比。NEB Monarch RNA清理套件始终优于其他市售的RNA清洁套件,从而从SGRNA合成反应中去除残留的NTP。

利用 CRISPR/Cas9 辅助基因组编辑体外调节马疱疹病毒 1 型复制动力学
摘要:(1)背景:马α疱疹病毒-1(EHV-1)是一种在全球大多数马群中流行的高度传染性病毒病原体。CRISPR/Cas9 等基因组编辑技术已成为精准 RNA 引导基因组修饰的有力工具;(2)方法:设计了单向导 RNA(sgRNA)以靶向三个必需基因(ORF30、ORF31 和 ORF7)和一个非必需基因(ORF74),并确定其对体外病毒复制动力学的影响;(3)结果:我们证明靶向必需溶解基因的 sgRNA 降低了 EHV-1 复制,而靶向 ORF74 的 sgRNA 的影响可忽略不计。靶向 ORF30 的 sgRNA 对抑制 EHV-1 复制表现出最强的效果,病毒基因组拷贝数和传染性子代病毒产量均有所减少。下一代测序鉴定出选择性sgRNA特定切割位点缺失的变异体,并评估了不同sgRNA之间的组合,发现靶向ORF30和ORF7的sgRNA双重组合与使用单个sgRNA相比,能显著抑制病毒复制至较低水平,表明存在协同效应;(4)结论:数据表明sgRNA引导的CRISPR/Cas9可用于体外抑制EHV-1复制,表明这种可编程技术可用于开发一种新颖、安全、有效的EHV-1治疗和预防方法。

sgRNA、启动子及外植体对芥兰CRISPR/Cas9系统基因编辑效率的影响
1 四川农业大学园艺学院,成都 611130;2021305054@stu.sicau.edu.cn(WH);zhengaihong@stu.sicau.edu.cn(AZ);hh820423@163.com(HH);2021205028@stu.sicau.edu.cn(XL);2022205029@stu.sicau.edu.cn(QL);201906195@stu.sicau.edu.cn(LL);2022205026@stu.sicau.edu.cn(RL);huangzhi@sicau.edu.cn(ZH);13185@sicau.edu.cn(YQ);13920@sicau.edu.cn(YT);10650@sicau.edu.cn(HL); zhangf@sicau.edu.cn (FZ) 2 遵义师范学院生物与农业技术学院,遵义 563006,浙江;czf810@163.com 3 毕节市农业科学研究所,毕节 551700,浙江;majie_011@126.com 4 浙江大学园艺系,杭州 310058,浙江 * 通讯地址:qmwang@zju.edu.cn (QW); bsun@sicau.edu.cn (BS); 电话:+86-571-88982278 (QW); +86-28-86291848 (BS) † 这些作者对本文贡献相同。

农业和生物技术-Purwati等。
生物技术应用具有基因工程方法,例如基因组编辑,以改善植物的性质,目的是提高结果的质量。CRISPR/CAS9成功地修改前所未有的精确度的基因组可能是由准确性,效率,成本效益和易用性引起的。CRISP/CAS9基因组编辑机制是通过插入,更换,去除一个或多个碱基的特定序列来操纵基因。由于基因的插入或变化,CRISPR/CAS方法会在基因组水平上具有破坏性的目标(脱离目标)之外的影响。本文讨论了CRISPR/CAS9,CRISPR/CAS9机制的开发以及在CRISPR/CAS9系统中经常发生的最小化方法。可以通过多种方式完成最小化攻击目标的方法,即:(1)SGRNA修饰与SGRNA GC含量,SGRNA长度,SGRNA长度,截断GRNA,SGRNA化学修饰,SGRNA化学修饰以及SILICO中SGRNA的修饰,(2)Cas Protein Modification和(3)CARS crispr crispr of CRRS PRERPR。


FEMS2023-摘要书.pdf
我们发现,与含有 8 个或更少典型 sgRNA(“sg0”-“sg8”)的样本(Ct:26.3,15.9 – 32.5)相比,含有全套 9 个典型 sgRNA(“sg9”)的样本与粗病毒载量(Ct:19.0,11.3 – 27.7)呈显著正相关(p ≤ 0.001)。sgORF7b 表达最少,在含有部分典型 sgRNA 的样本中 96.7% 未检测到。含有“sg9”模式的样本收集较早(早期 63.3% vs 晚期 27.4%)。9 个典型 sgRNA 的检测率变化与粗病毒载量和取样日相关,但与年龄、性别和肺炎无关。接收者操作特性 (ROC) 曲线分析表明,检测全套 9 个典型 sgRNA(AUC = 0.91,95% CI 0.88-0.94)、sgRNA ORF7b(0.90,0.87 – 0.93)和 sgORF7a(0.89,0.84 – 0.93)与 BA.2 和 D614G 变体的粗病毒载量表现出最佳关联。
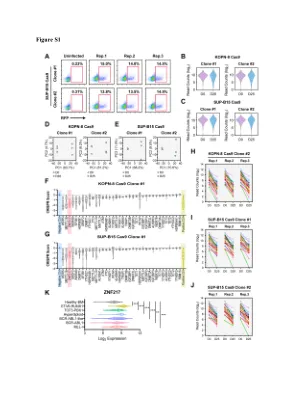
图S1
(a)使用SUP-B15 Cas9单克隆,SGRNA库的慢病毒转导效率。(b)使用KOPN-8 CAS9单个克隆在CRISPR屏幕的“初始”和“最终”点收集的NGS样品中SGRNA读数的分布。(c)使用SUP-B15 Cas9单个克隆在CRISPR屏幕的“初始”和“最终”点收集的NGS样品中SGRNA读数的分布。(d)使用KOPN-8 CAS9单个克隆在CRISPR屏幕上收集的NGS样品的PCA分析。(E)使用SUP-B15 Cas9单个克隆在CRISPR屏幕的“初始”和“最终”点收集的NGS样品的PCA分析。(f)使用KOPN-8 CAS9单个克隆,针对CRISPR屏幕中36个RNA和DNA甲基化机械基因的SGRNA的CRISPR得分。CRISPR得分已针对阴性对照SGRNA的平均得分进行标准化(设置为0.0)。(g)使用SUP-B15 Cas9单克隆,针对CRISPR筛选中36个RNA和DNA甲基化机械基因的SGRNA的CRISPR得分。CRISPR得分已针对阴性对照SGRNA的平均得分进行标准化(设置为0.0)。(h)在KOPN-8 CAS9克隆#2中靶向Znf217的25个SGRNA的计数。(i)读取针对SUP-B15 Cas9克隆#1中Znf217的25个SGRNA的计数。(j)读取25个针对Znf217的SGRNA的计数,SUP-B15 Cas9克隆#2。(k)ZnF217在不同的B-ALL亚型和健康的骨髓中的表达。Znf217表达数据来自白血病(MILE)研究的微阵列创新(登录GSE13159)。n = 70,MLL-R; BCR-ABL1 n = 122; n = 237,类似于bcr- abl1; n = 40用于高二倍体; TCF3-PBx1的n = 36; ETV6-RUNX1的n = 58; n = 73用于健康的BM。使用两尾t检验计算p值。** p <0.01; *** p <0.001。