XiaoMi-AI文件搜索系统
World File Search System自噬

β-氯环己烷触发神经炎症活性,表观遗传组蛋白翻译后修饰和认知功能障碍
与年龄相关的肌肉干细胞(MUSC)再生能力的减少与细胞自主和非细胞自主变化有关,这是由于全身和骨骼肌环境改变而导致的,最终导致MUSC数量和功能下降。先前的研究表明,通过在激活的MUSC中进行自噬,STAT3在损伤激活再生后驱动MUSC扩张和分化方面起着关键作用。然而,自噬在寿命中逐渐下降,并导致MUSC介导的老年肌肉再生受损。在这里,我们表明STAT3抑制作用恢复了老年MUSC的自噬过程,从而恢复了MUSC促进老年小鼠肌肉再生的能力。我们表明,通过促进自噬相关基因的转录以及在细胞质水平的转录,可以通过靶向EIF2α的STAT3/PKR磷酸化来激活核水平的自噬。这些结果表明STAT3 Inhi-Bition是一种潜在的干预措施,以扭转与年龄相关的自噬块,从而破坏MUSC再生肌肉的能力。他们还揭示了STAT3通过转录依赖性和独立的自噬调节来调节MUSC功能。

靶向 Beclin 1 的钉合肽干扰自噬可诱导线粒体应激并抑制胰腺癌细胞增殖
简单总结:长期以来,自噬被认为在包括 PDAC 在内的多种癌症中发挥促增殖和抗增殖作用。由于自噬抑制剂 CQ 在 PDAC 临床试验中未能显示出治疗效果,因此值得探索替代方法(即提高自噬活性)是否可以发挥抗肿瘤作用。我们的研究旨在评估 Beclin 1 靶向钉合肽是否可以通过扰乱已经升高的自噬过程在 PDAC 中发挥抗增殖作用。我们的研究首次报告了 Beclin 1 靶向钉合肽 Tat-SP4 通过过度自噬、增强的 EGFR 内溶酶体降解和显著的线粒体应激的综合作用有效抑制 PDAC 细胞的增殖。Tat-SP4 在 PDAC 细胞中诱导非凋亡性细胞死亡,这与 CQ 诱导的细胞凋亡形成鲜明对比。总之,Tat-SP4 对自噬过程的干扰可能成为 PDAC 的一种新治疗方法。

稀有疾病治疗的前景
罕见的疾病的特征是其发病率低,但是有多种类型的罕见疾病导致大量患有全球罕见疾病的人。尽管到目前为止罕见疾病的治疗方面已经取得了一些进展,但遭受个人罕见疾病的小人群的地理分散,与孤儿药物开发相关的高成本以及其他因素挑战了这些疾病的临床治疗剂的发展。自噬是真核细胞中高度保守的降解过程,对于维持细胞稳态至关重要。研究表明,自噬的失调促进了许多罕见疾病的病理,例如VICI综合征,Danon病和间皮瘤。对自噬如何参与稀有疾病的方式有更清晰的理解可以帮助开发新的治疗方法。在这篇评论中,我们将简要介绍自噬,然后重点介绍稀有疾病和自噬之间的联系。还将讨论针对自噬进行稀有疾病治疗的前景和挑战。

AMPK在抑制自噬和长期氨基酸剥夺作者中MTORC1信号的重新激活中的意外作用
摘要AMPK促进分解代谢并抑制合成代谢的细胞代谢,以在能量应激期间促进细胞存活,部分通过抑制MTORC1,这是一种合成代谢激酶,需要足够水平的氨基酸。我们发现缺乏AMPK的细胞显示出在氨基酸剥夺长期导致的营养应激期间凋亡细胞死亡增加。我们假定自噬受损解释了这种表型,因为一种普遍的观点认为AMPK通过ULK1的磷酸化启动了自噬(通常是亲生响应)。出乎意料的是,在缺乏AMPK的细胞中,自噬仍然没有受损,正如多个细胞系中的几个自噬读数所监测的那样。更令人惊讶的是,在氨基酸剥夺期间,不存在AMPK的ULK1信号传导和LC3B脂质增加,而AMPK介导的ULK1 S555的磷酸化(拟议启动自噬的站点)在氨基酸戒断或药理学MTORC1抑制后降低了ULK1 S555(拟议启动自噬)的磷酸化。此外,用化合物991,葡萄糖剥夺或氨基酸戒断引起的AICAR钝化自噬的AMPK激活。这些结果表明AMPK激活和葡萄糖剥夺抑制自噬。作为AMPK控制的自噬在意外方向上,我们检查了AMPK如何控制MTORC1信号传导。矛盾的是,我们观察到在长时间氨基酸剥夺后缺乏AMPK的细胞中MTORC1的重新激活受损。这些结果共同反对既定的观点,即AMPK促进自噬并普遍抑制MTORC1。这些发现促使对AMPK及其对自噬和MTORC1的控制如何影响健康和疾病进行了重新评估。此外,在延长氨基酸剥夺的背景下,它们揭示了AMPK在抑制自噬和MTORC1信号传导中的意外作用。关键字:mtor; S6K1; 4EBP1; lc3b; ULK1; ATG16L1;化合物991;葡萄糖剥夺; aicar;细胞存活缩写:AAS:氨基酸; ADP:双磷酸腺苷; AICAR:5-氨基咪唑-4-羧酰胺核糖核苷酸; AMP:单磷酸腺苷; AMPK:AMP激活的蛋白激酶; ATG14:自噬相关14; ATG16L1:自噬相关16,如1; ATG5:自噬相关5; BAFA1:Bafilomycin A1; DKD:双重击倒; DKO:双淘汰赛; ECL:增强的化学发光; LC3B:微管相关蛋白1A/1B轻链3B; MEF:小鼠胚胎成纤维细胞; MTORC1:雷帕霉素复合物1的机械靶标; MTORC2:雷帕霉素复合物2的机械靶标; p62:泛素结合蛋白p62,又名SQSTM1/secestosoms 1; S6K1核糖体蛋白S6激酶1; 4EBP1,EIF4E [真核起始因子4E]结合蛋白1; TEM:透射电子显微镜; ULK1:UNC-51样激酶1; VPS34,液泡蛋白排序34。

全基因组 - crispr-screen-识别tmem41b-as-a-gene ...
大自源性是一个细胞内降解过程,需要多个自噬相关(ATG)基因。在这项研究中,我们使用自噬型号报告基因GFP-LC3-RFP进行了全基因组筛选,并鉴定出TMEM41B作为一种新型ATG基因。TMEM41B是一种位于内质网(ER)中的多层膜蛋白。它在液泡膜蛋白1(VMP1)中也发现了一个保守的结构域,这是另一种ER多跨度膜蛋白,对于自噬,酵母菌TVP38必不可少的,以及推定的半转生蛋白的细菌deda家族。TMEM41B的缺失阻止了早期的自噬体的形成,从而导致ATG蛋白和小囊泡的积累,但不会拉长自噬体样结构。此外,在TMEM41B -KNOCKOUT(KO)细胞中积累的脂质液滴。TE表型类似于VMP1 -KO细胞的表型。的确,TMEM41B和VMP1在体内和体外形成了复杂的复杂,VMP1的过表达恢复了TMEM41B -KO细胞中的自噬量。TESE结果表明,TMEM41B和VMP1在自噬体形成的早期步骤中起作用。
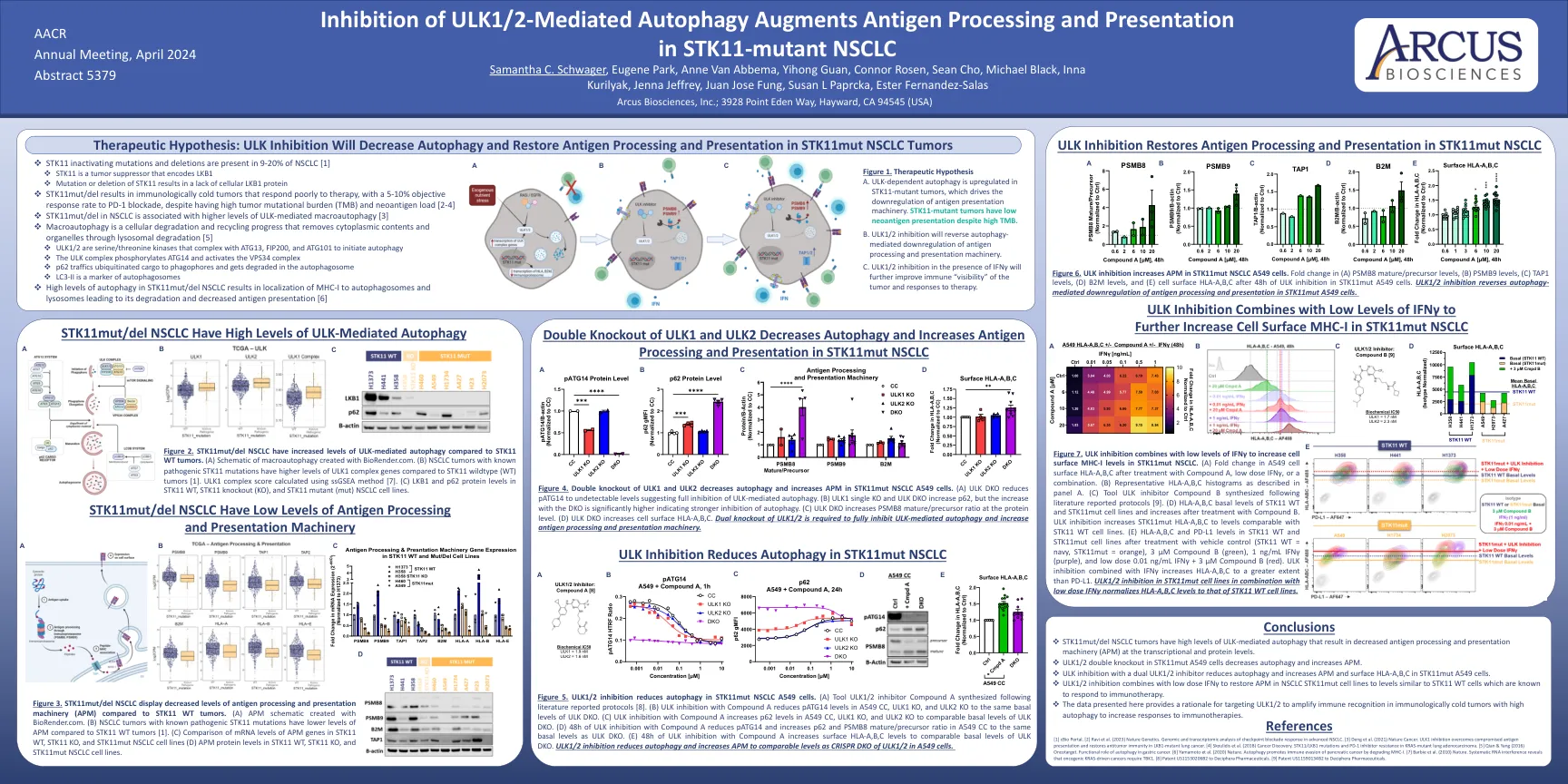
抑制ULK1/2介导的自噬增强抗原加工和表现 ARC-9:一项评估基于伊特鲁马德的治疗组合的随机研究
图2。与STK11 WT肿瘤相比, STK11MUT/DEL NSCLC具有ULK介导的自噬水平升高。 (a)用biorender.com创建的大噬细胞的示意图。 与STK11野生型(WT)肿瘤相比,具有已知致病性STK11突变的NSCLC肿瘤具有更高水平的ULK1复合基因[1]。 ULK1复合分数使用SSGSEA方法计算[7]。 (C)STK11 WT,STK11敲除(KO)和STK11突变体(MUT)NSCLC细胞系中的LKB1和P62蛋白水平。 图4。 ULK1和ULK2的双重敲除可降低自噬并增加STK11MUT NSCLC A549细胞中的APM。 (a)ULK DKO将PATG14降低至无法检测的水平,表明对ULK介导的自噬完全抑制。 (b)ULK1单个KO和ULK DKO增加了p62,但随着DKO的增加,dKO的增加表明自噬抑制更强。 (c)ULK DKO在蛋白质水平上增加了PSMB8成熟/前体比率。 (d)ULK DKO增加了细胞表面HLA-A,b,c。 需要ULK1/2的双重敲除以完全抑制ULK介导的自噬并增加抗原加工和表现机制。STK11MUT/DEL NSCLC具有ULK介导的自噬水平升高。(a)用biorender.com创建的大噬细胞的示意图。与STK11野生型(WT)肿瘤相比,具有已知致病性STK11突变的NSCLC肿瘤具有更高水平的ULK1复合基因[1]。ULK1复合分数使用SSGSEA方法计算[7]。 (C)STK11 WT,STK11敲除(KO)和STK11突变体(MUT)NSCLC细胞系中的LKB1和P62蛋白水平。 图4。 ULK1和ULK2的双重敲除可降低自噬并增加STK11MUT NSCLC A549细胞中的APM。 (a)ULK DKO将PATG14降低至无法检测的水平,表明对ULK介导的自噬完全抑制。 (b)ULK1单个KO和ULK DKO增加了p62,但随着DKO的增加,dKO的增加表明自噬抑制更强。 (c)ULK DKO在蛋白质水平上增加了PSMB8成熟/前体比率。 (d)ULK DKO增加了细胞表面HLA-A,b,c。 需要ULK1/2的双重敲除以完全抑制ULK介导的自噬并增加抗原加工和表现机制。ULK1复合分数使用SSGSEA方法计算[7]。(C)STK11 WT,STK11敲除(KO)和STK11突变体(MUT)NSCLC细胞系中的LKB1和P62蛋白水平。图4。ULK1和ULK2的双重敲除可降低自噬并增加STK11MUT NSCLC A549细胞中的APM。(a)ULK DKO将PATG14降低至无法检测的水平,表明对ULK介导的自噬完全抑制。(b)ULK1单个KO和ULK DKO增加了p62,但随着DKO的增加,dKO的增加表明自噬抑制更强。(c)ULK DKO在蛋白质水平上增加了PSMB8成熟/前体比率。(d)ULK DKO增加了细胞表面HLA-A,b,c。需要ULK1/2的双重敲除以完全抑制ULK介导的自噬并增加抗原加工和表现机制。

溶酶体存储,高彻和帕金森氏病的自噬和先天免疫受损:药物发现的见解
自噬 - 溶酶体途径的损害越来越涉及帕金森氏病(PD)。GBA1突变引起溶酶体储存障碍Gaucher病(GD),是PD的最常见遗传危险因素。GBA1突变已显示会引起自噬 - 溶酶体损伤。 不良细胞成分的自噬降解有缺陷与多种病理有关,包括正常蛋白质稳态的丧失,特别是α-突触核蛋白和先天免疫功能障碍。 在PD和GD中观察到后者。 在这里,我们将讨论自噬和免疫失调之间的机理联系,以及这些病理学在肠道和大脑之间在这些疾病中的沟通中的可能作用。 在神经性GD(NGD)的蝇模型中的最新工作显示肠自噬缺陷导致胃肠道功能障碍和免疫激活。 雷帕霉素治疗部分逆转了自噬阻滞并降低了免疫活性,与生存率增加并改善了运动能力。 肠道微生物组的改变是神经炎症的关键驱动力,研究表明,在NGD蝇中消除了微生物组,而PD的小鼠模型可以改善脑部炎症。 在这些观察结果之后,将溶酶体 - 自噬途径,先天免疫信号传导和微生物组营养不良症讨论为PD和GD中的潜在治疗靶标。 本文是讨论会议问题的一部分,“理解神经变性中的内聚糖网络”。GBA1突变已显示会引起自噬 - 溶酶体损伤。不良细胞成分的自噬降解有缺陷与多种病理有关,包括正常蛋白质稳态的丧失,特别是α-突触核蛋白和先天免疫功能障碍。在PD和GD中观察到后者。在这里,我们将讨论自噬和免疫失调之间的机理联系,以及这些病理学在肠道和大脑之间在这些疾病中的沟通中的可能作用。在神经性GD(NGD)的蝇模型中的最新工作显示肠自噬缺陷导致胃肠道功能障碍和免疫激活。雷帕霉素治疗部分逆转了自噬阻滞并降低了免疫活性,与生存率增加并改善了运动能力。肠道微生物组的改变是神经炎症的关键驱动力,研究表明,在NGD蝇中消除了微生物组,而PD的小鼠模型可以改善脑部炎症。在这些观察结果之后,将溶酶体 - 自噬途径,先天免疫信号传导和微生物组营养不良症讨论为PD和GD中的潜在治疗靶标。本文是讨论会议问题的一部分,“理解神经变性中的内聚糖网络”。

多激酶抑制剂AD80在胰腺癌细胞中诱导有丝分裂灾难和自噬
1个在Onco-Muno-Herasology(LIM-31)的发病机理和靶向疗法的医学研究实验室(LIM-31),内科系,血液学部,ST-o Paulo University faculdade de Medicina,SãoPaulo,SãoPaulo 01246-903,巴西; kelilima@usp.br(K.L.); marianenasc@hotmail.com(m.c.d.n.); edumrego@hotmail.com(E.M.R.)2生物医学科学研究所,萨尔·保罗大学,巴西sâo paulo 05508-000; liviamirands@usp.br(L.B.L.D.M.); bruolialmeida@usp.br(B.O.D.A.); drguilhermealcantara.usp@gmail.com(g.a.s.a.)3生物医学研究所的细胞和发育生物学系,萨尔·保罗大学,巴西s-o paulo 05508-000; anabega@usp.br(A.D.M.B.G.); glaucia.usp@gmail.com(G.M.M.-S。) *通信:jamachadoneto@usp.br;电话。: +55-11-3091-7467

高血糖通过 PI3Kγ 依赖的自噬缺陷加剧平滑肌泡沫细胞的形成
高血糖通过 PI3Kγ 依赖的缺陷自噬加剧平滑肌泡沫细胞的形成 Labrana H 1* ., Wahart A 1* ., Cormier K 1 ., Solinhac R 1 ., Swiader A 1 ., Mentouri I 1 ., Smirnova N 1 ., Malet N 1 ., Gayral S 1 ., Ramel D 1 ., Auge N 1 **., Laffargue M 1 ** 1 I2MC,法国国家健康与医学研究中心 (INSERM) U1297,法国 *,** 同等贡献

自噬抑制剂3-甲基趋化对糖尿病小鼠模型的影响
摘要●目的:研究自噬抑制剂3-甲基趋化(3-MA)在糖尿病小鼠模型(DM)和潜在机制上的作用。●方法:将雄性C57BL/6J小鼠随机分为正常对照组(NC组)和DM组。dm是通过多种低剂量腹膜内注射链蛋白酶(STZ)60 mg/kg●连续5天诱导的。dm小鼠随机细分为未处理的组(DM组),3-ma(10 mg/kg●dm gavage)治疗组(DM+3-ma组)和氯喹(CQ; 50 mg/kg通过腹膜内注射)治疗组(DM+CQ组)。每周记录空腹血糖(FBG)水平。在实验结束时,收集了视网膜样品。The expression levels of pro-apoptotic proteins cleaved caspase-3, cleaved poly ADP-ribose polymerase 1 (PARP1) and Bax, anti-apoptotic protein Bcl-2, fibrosis- associated proteins Fibronectin and type 1 collagen α1 chain (COL1A1), vascular endothelial growth factor (VEGF), inflammatory factors interleukin (IL)-1β和肿瘤坏死因子(TNF)-α以及自噬相关蛋白LC3,