XiaoMi-AI文件搜索系统
World File Search System裂解物

使用 3M Harvest RC 色谱澄清剂从 HEK 洗涤剂裂解物中去除宿主细胞 DNA 的调查案例研究
腺相关病毒 (AAV) 是广泛用于递送基因疗法以治疗各种人类疾病的载体。截至 2024 年 3 月,美国 FDA(食品和药物管理局)已批准了 5 种基于 AAV 的疗法,并且正在进行 200 多项临床试验。1 生产 AAV 的方法包括在宿主细胞(例如 HEK293)中进行瞬时三重转染、基于杆状病毒的表达系统和无辅助病毒方法(例如单纯疱疹病毒 (HSV) 系统)。2 AAV 颗粒通常在细胞内,AAV 纯化过程通常始于通过洗涤剂裂解将病毒从宿主细胞中释放出来;该细胞裂解过程也经常将 hcDNA 释放到液体中。最终产品中存在 hcDNA 对经过处理的细胞构成了重大的安全隐患

质粒DNA maxiprep套件产品插件
NORGEN的纯化技术纯化基于自旋色谱柱色谱法。优先从其他细胞成分(例如基因组DNA和RNA)中纯化质粒DNA。该过程涉及首先颗粒的隔夜细菌培养物具有质粒DNA(请参阅第3页的流程图)。然后,使用提供的重悬溶液AZ重悬细菌颗粒,并使用裂解缓冲液N进行细菌。然后添加缓冲液。然后添加缓冲液TN,从而导致溶液中存在的基因组DNA和蛋白质。然后通过离心阐明裂解物,以去除含有质粒DNA的裂解物中的沉淀蛋白和基因组DNA。然后将澄清的裂解物加载到自旋柱上。Norgen的柱以取决于离子浓度的方式结合DNA,因此,只有质粒DNA才能与柱结合,而大多数消化的RNA和蛋白质将在流动中除去或保留在柱床的顶部。然后用提供的洗涤溶液J洗涤结合的DNA,以去除剩余的杂质。最后,无内毒素纯化的质粒DNA用洗脱缓冲液洗脱。纯化的DNA是最高质量的,可用于许多下游应用,包括测序,克隆和转染。

热休克黑色素瘤细胞裂解物疫苗通过原型效应 T 细胞抑制肿瘤生长来增强肿瘤浸润
摘要 背景 免疫检查点阻断剂 (ICB) 疗法已显示出对某些癌症患者的生存益处。然而,许多患者仍然对治疗有抵抗力或获得性耐药性,这促使人们探索补充免疫疗法。因此,癌症疫苗提供了一种有吸引力的替代方案。多种肿瘤相关抗原与强效佐剂的最佳递送似乎对疫苗有效性至关重要。 方法 这里,使用 B16F10 小鼠黑色素瘤模型测试了一种名为 TRIMELVax 的仿制黑色素瘤疫苗原型。该疫苗由热休克处理的肿瘤细胞裂解物与 Concholepas concholepas 血蓝蛋白作为佐剂制成。结果虽然 B16F10 裂解物提供了适当的黑色素瘤相关抗原,但通用人类黑色素瘤细胞裂解物和血蓝蛋白佐剂均能发出危险信号,促进常规 1 型树突状细胞 (cDC1) 的激活、吞噬作用和有效的抗原交叉呈递。TRIMELVax 抑制肿瘤生长并提高小鼠存活率,诱导细胞和体液免疫反应。此外,这种疫苗产生的肿瘤内 cDC1 频率增加,但没有产生常规 2 型树突状细胞 (cDC2)。与抗程序性细胞死亡蛋白 1 (PD-1) 单一疗法相比,还观察到 CD3 +、CD4 + 和 CD8 + T 细胞的浸润增强,而 TRIMELVax/抗 PD-1 组合产生更高的 CD4 + T 细胞肿瘤浸润。此外,TRIMELVax 促进了肿瘤中 PD-1 lo CD8 + T 细胞比例的增加,这是一种与肿瘤生长控制所需的原型效应细胞相关的表型,可防止功能失调的 T 细胞积聚。结论治疗性疫苗 TRIMELVax 可有效控制弱免疫原性和侵袭性的 B16F10 黑色素瘤肿瘤生长,即使在没有 ICB 的情况下也能延长荷瘤小鼠的生存期。TRIMELVax 表现出的强免疫原性鼓励对黑色素瘤患者进行临床研究。
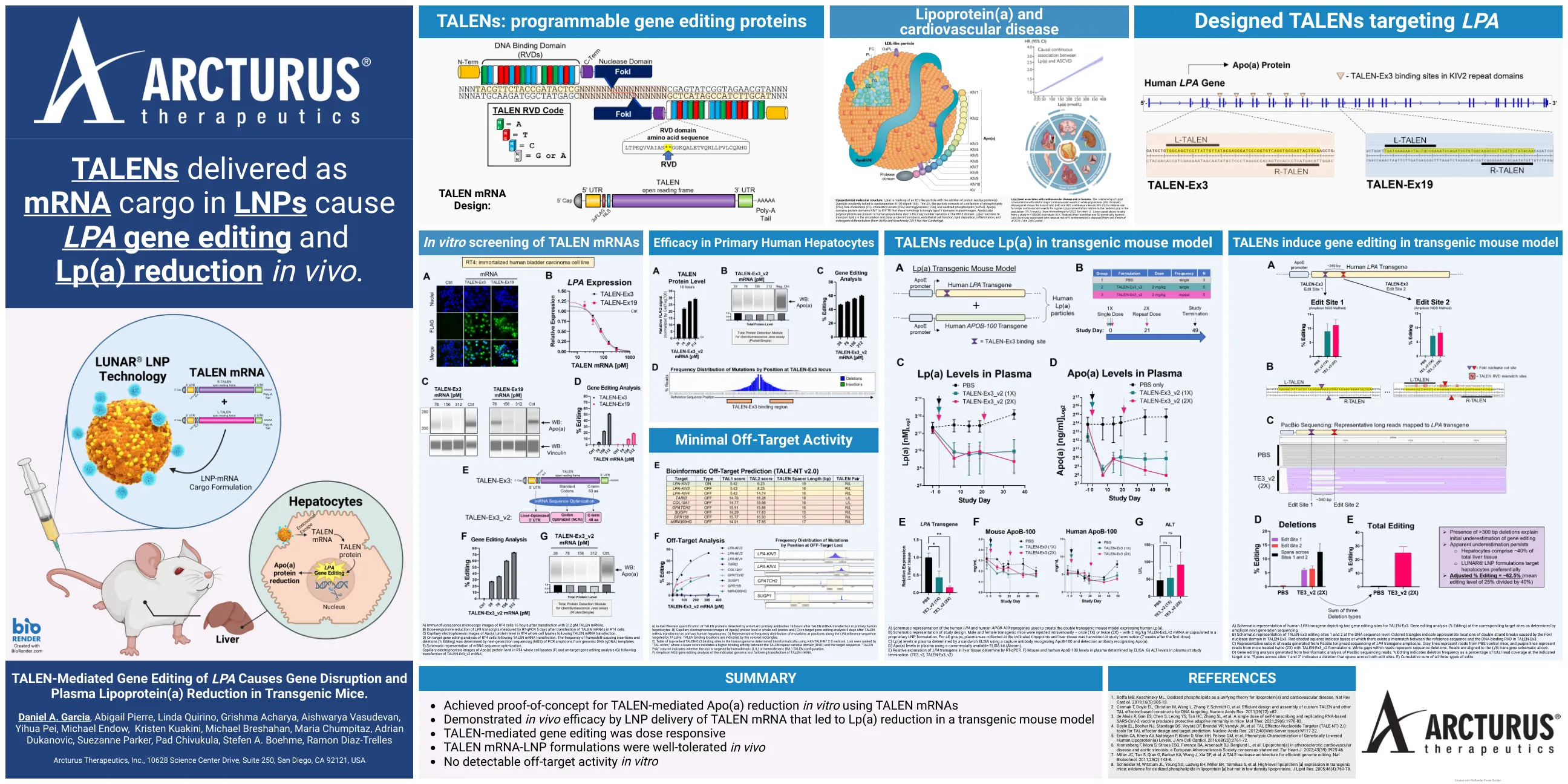
LPA 海报 ASGCT 2023
A) RT4 细胞转染 312 pM TALEN mRNA 16 小时后的免疫荧光显微镜图像。B) RT4 细胞转染 TALEN mRNA 5 天后,通过 RT-qPCR 测量 LPA 转录本的剂量反应性减少。C) TALEN mRNA 转染后 RT4 全细胞裂解物中 Apo(a) 蛋白水平的毛细管电泳图像。D) TALEN mRNA 转染后 RT4 细胞的靶向基因编辑分析。通过基因组 DNA (gDNA) 模板的 PCR 扩增子的下一代测序 (NGS) 确定导致移码的插入和缺失的频率 (编辑百分比)。E) mRNA 序列优化的示意图。转染 TALEN-Ex3_v2 mRNA 后 RT4 全细胞裂解物中 Apo(a) 蛋白水平的毛细管电泳图像 (F) 和靶向基因编辑分析 (G)。

最大化吞吐量的定量西方结果
(a)上面显示的是来自单个LEO运行的数据。使用重组GFP蛋白的8点标准曲线在弹药筒上三次。墨盒2、3和4用于定量来自2种不同批次和各种剂量的转导细胞裂解物中的GFP表达。重组蛋白以2.5 ng/ml的速度作为校准器在所有墨盒上作为用于墨盒校正的校准器。(b)上面显示的是来自单个JES运行的数据。使用重组GFP蛋白的8点标准曲线运行,并用于定量来自2个不同批次转导的细胞裂解物中的GFP表达。总体而言,每杰西斯运行的毛细血管数量有限,只能容纳标准曲线的一个复制和有限数量的样品。在杰西系统上处理96个样本至少需要4次运行和12小时。

无细胞蛋白质合成
摘要 蛋白质是药物靶标的主要来源,其中一些蛋白质本身就具有治疗潜力。其中,膜蛋白约占主要药物靶标的 50%。在药物发现过程中,以简单的方式高质量地快速生产不同类别的蛋白质的方法对于结构和功能分析非常重要。无细胞系统因其灵活性而不受任何细胞膜限制,正在成为生产蛋白质的一种有吸引力的替代方案。在生物生产环境中,基于来自不同来源的细胞裂解物且批次间一致的开放系统已成为无细胞合成目标蛋白质的催化剂。最重要的是,蛋白质可以加工用于下游应用,如纯化和功能分析,而无需转染、选择和扩增克隆。在过去的 5 年里,来自多种生物体的新型无细胞裂解物的可用性不断增加,它们用于合成多种蛋白质。尽管取得了这些进展,但在可扩展性、成本效益、蛋白质折叠和功能性方面仍然存在重大挑战。在本综述中,我们概述了来自不同来源的不同无细胞系统及其在生产各种蛋白质中的应用。此外,本文还讨论了中国仓鼠卵巢和 Sf 21 裂解物中含有内源性易位活性微粒体的无细胞系统在膜蛋白合成方面的一些最新进展。我们特别强调了内部核糖体进入位点序列在更有效地生产蛋白质方面的应用,以及位点特异性掺入非规范氨基酸对标记应用和使用无细胞系统创建抗体药物偶联物的重要性。我们还讨论了克服从实验室层面将无细胞平台商业化以用于未来药物开发的主要挑战的策略。
人类 AXIN-2 全检测试剂盒
试剂盒特异性:此检测试剂盒含有可识别 AXIN-2 上不同表位的抗体。此试剂盒检测的蛋白质对应于 UniProt ID Q9Y2T1。AXIN-2 也称为轴蛋白样蛋白 (Axil)、轴抑制蛋白 2 和 Conductin。这些抗体可识别人类来源的 AXIN-2。其他物种应根据具体情况进行测试。对照裂解物信息:阳性对照裂解物:由 SW 48 细胞制备,在含有 10% FBS 的培养基中在 T175 烧瓶中培养至汇合,并用 2.5 mL 裂解缓冲液裂解。代表性数据:使用 2 板 2 孵育方案获得的数据。将 SW 48 细胞以 40K 细胞/孔接种在 96 孔板中并孵育过夜。用指定浓度的 Tankyrase 抑制剂 (XAV-939) 处理细胞 24 小时。使用裂解缓冲液裂解细胞,然后使用相应的 SureFire Ultra 试剂盒分别测定 AXIN-2 Total 和 ERK1/2 Total。相当于约 4,000 个细胞/数据点。

核纳质质粒DNA纯化用户手册
NucleOmag®质粒程序利用了修饰的碱性裂解方案,并结合了适当的缓冲条件下核酸对顺磁珠的可逆吸附。颗粒细菌被重悬于缓冲液A1中。质粒DNA通过裂解缓冲液A2从细胞中解放出来,然后使用缓冲液S3进行中和和沉淀。粗裂解物可以通过离心或使用NucleOmag®清除珠(用于裂解液清除的专门的顺磁珠)清除。用于将核酸与顺磁珠,结合缓冲液和核瘤®M珠结合的结合添加到清除的裂解物中。磁分离后,通过获得专利的排毒缓冲液ERB去除内毒素和蛋白质。用洗涤缓冲液和空气干燥除去盐或残留乙醇等进一步的污染物。纯质粒DNA用低盐洗脱缓冲液或水洗脱,并准备好用于任何常见的下游应用(包括转染)(仅研究)。核对®质粒试剂盒已设计用于自动磁杆系统。


干细胞研究-Chen Lab-休斯顿大学
分子超分辨率显微镜(Chen等,2015),Geneti Cally用SNAP-TAG(一种突变的人O 6-烷基鸟嘌呤-DNA-烷基转移酶)标记了SOD1,可以用各种合成探测器将其共价标记(Keppler et al。通过两轮CRISPR-CAS9介导的同源性建议,获得了遗传修饰的H1 HESC纯合子克隆(H1_SOD1-SNAP)。在三氨基 - 酸 - 酸 - 链接中,将SNAP基因插入了SOD1编码区的C末端(图1 a)。H1 HESC与单个诱导RNA(SGRNA)-CAS9 PLAS MIDS和含有质粒的重组供体共转染。挑选了在分裂克隆中出现的并进行筛选以进行快照插入。首先获得了由一个快照敲入等位基因组成的杂合子克隆,并进行了第二轮CRISPR-CAS9诱导的SNAP敲击。然后,我们成功地产生了通过基因分型PCR,Sanger测序和Southern blot分析确认的纯合子快照敲入克隆(图1 B,补充。 图 1和补充。 图 2)。 通过使用抗SNAP和抗SOD1抗体进行免疫印迹证实了SOD1-SNAP融合蛋白在敲门细胞裂解物中的表达(图1 B,补充。图1和补充。图2)。通过使用抗SNAP和抗SOD1抗体进行免疫印迹证实了SOD1-SNAP融合蛋白在敲门细胞裂解物中的表达(图1 F和补充。图4)。大多数SOD1-SNAP蛋白在敲击细胞裂解物中保持完整,仅几乎无法检测到的未标记的SOD1信号水平,这可能源自裂解(图1 f)。与H1亲本细胞中的内源SOD1水平相比,总体SOD1-SNAP蛋白水平降低了,这表明TAG