XiaoMi-AI文件搜索系统
World File Search System骨细胞

全骨韧性与ZDSD 2型糖尿病大鼠模型中的运河和骨细胞缺陷有关
2型糖尿病(T2DM)与骨骼质量无关的骨折风险增加有关。这种增加的骨折风险的确切起源仍未得到充分理解。使用多基因糖尿病大鼠模型,同步辐射微型计算层析成像(SR µ CT)以及原位扫描电子显微镜(SEM)断裂韧性,我们将显微镜的变化与糖尿病比股骨的韧性和材料特性相关联。糖尿病大鼠模型(ZDSD)显示出隔夜禁食高血糖和增加年龄的含量。此外,我们测量了糖尿病大鼠中产物后特性和韧性的损害。在该ZDSD模型中也影响了皮质几何形状和孔隙率。我们测量了与lacunar体积减少相关的骨细胞lacunar密度的降低。此外,我们发现运河密度降低,同时保持类似的管直径。这些结果表明糖尿病会损害骨骼重塑,从而影响骨骼微观。由于运河和空隙也与外在的韧性机制有关,因此我们将韧性下降归因于这些微观结构的变化。总而言之,我们表明lacunae和运河密度的变化以及年龄的积累,降低了T2DM大鼠骨的韧性。
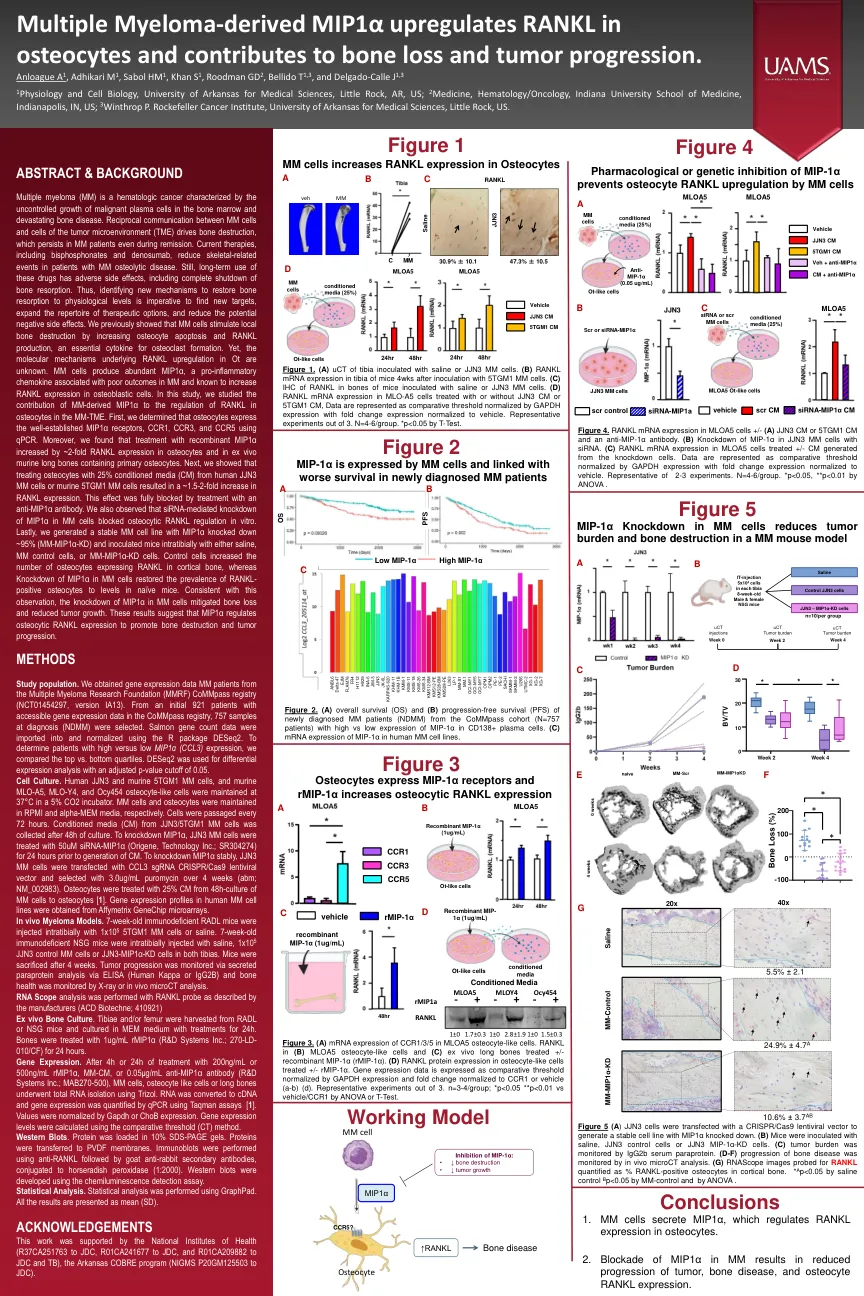
多发性骨髓瘤的MIP1α上调RANKL ...
多发性骨髓瘤(MM)是一种血液学癌,其特征是骨髓和毁灭性骨病中恶性血浆细胞的生长不受控制。MM细胞与肿瘤微环境(TME)细胞之间的相互通信驱动骨骼破坏,即使在缓解过程中,MM患者也持续存在。当前的疗法,包括双膦酸盐和denosumab,减少了MM骨化疾病患者的骨骼相关事件。仍然,这些药物的长期使用具有不利的副作用,包括骨吸收的完全关闭。因此,必须确定新的机制以将骨吸收恢复到生理水平,这对于寻找新靶标,扩展治疗选择的曲目并减少潜在的负面副作用至关重要。我们先前表明,MM细胞通过增加骨细胞凋亡和RANKL产生来刺激局部骨破坏,这是破骨细胞形成的必不可少的细胞因子。然而,OT中RANKL上调的分子机制尚不清楚。mM细胞产生丰富的MIP1α,这是一种与MM结局不佳相关的促炎性趋化因子,并且已知会增加成骨细胞中RANKL表达。在这项研究中,我们研究了MM衍生的MIP1α对MM-TME中骨细胞中RANKL调节的贡献。首先,我们确定骨细胞使用qPCR表达了良好的MIP1α受体CCR1,CCR3和CCR5。此外,我们发现重组MIP1α的治疗在骨细胞和含有原发性骨细胞的离体鼠长骨中增加了约2倍的RANKL表达。接下来,我们表明,从人JJN3 MM细胞或鼠5TGM1 MM细胞中使用25%条件培养基(CM)处理骨细胞,RANKL表达增加了约1.5-2倍。用抗MIP1α抗体处理完全阻断了这种作用。我们还观察到siRNA介导的MM细胞中MIP1α的敲低阻断了体外骨细胞RANKL调节。最后,我们生成了一个稳定的MM细胞系,MIP1α击倒了〜95%(MM-MIP1α-KD),并用盐水,MM对照细胞或MM-MIP1α-KD细胞在室内接种小鼠。对照细胞增加了在皮质骨中表达RANKL的骨细胞的数量,而MM细胞中MIP1α的敲低使RANKL-阳性骨细胞的患病率恢复为幼稚小鼠的水平。与该观察结果一致,MM细胞中MIP1α的敲低减轻骨质流失和肿瘤生长减少。这些结果表明MIP1α调节骨细胞RANKL表达以促进骨骼破坏和肿瘤进展。

2.5临床概述 div>
如图2所示,骨骼重塑,骨骼在成年骨骼中不断重塑,这是通过骨质化的破骨细胞和形成骨成骨细胞的协调和顺序作用。这些细胞起作用可修复微塑料并适应骨骼结构满足机械和代谢需求。骨细胞>占所有骨细胞的95%,调节骨骼重塑。成骨细胞源自间充质干细胞(MSC),专门产生细胞外骨基质,包括I型胶原蛋白和非胶原蛋白,包括骨环钙蛋白,骨tec蛋白,骨修蛋白和骨4。随后通过沉积羟基磷灰石的沉积将骨基质矿化和僵硬。人体钙的约95%掺入骨基质中。破骨细胞源自巨型和单核细胞谱系的造血干细胞(HSC)。从前体细胞向活化的多核细胞的分化至关重要地取决于作用于整骨蛋白等级的核因子kappa b(rank)配体的受体激活剂(rankL),以及巨噬细胞刺激性刺激因子(M-CSF)的允许水平。RANKL主要由成骨细胞谱系细胞(MSC,成骨细胞和成骨细胞)和淋巴细胞产生。成熟的骨 - 分辨破骨细胞是大型多核细胞。使用密封区在骨表面附着并用褶皱的边框增强其表面,成熟的破骨细胞分泌盐酸(HCL)创建一种酸性微环境,其中诸如calterepsin k之类的酶(例如canterpsin k),降低了I型I型collagen collagen,是最活跃的(21,73,73,85)。

药物相关性颌骨坏死中的免疫功能障碍
摘要:药物相关性颌骨坏死 (MRONJ) 的发病机制是多因素的,人们普遍认为抗吸收药物 (ARD),包括双膦酸盐 (BP) 和地舒单抗 (Dmab),是主要决定因素之一。这些药物的暴露时间、累积剂量和给药强度是治疗患者时需要考虑的关键参数,因为癌症患者的 MRONJ 发病率最高。BPs 和 Dmab 对骨骼的作用机制不同,但它们对与骨细胞相互作用的免疫亚群也发挥不同的影响,从而导致 MRONJ 的发生。在这里,我们总结了 ARDs 对不同免疫细胞亚群的主要影响,从而影响骨细胞,特别是破骨细胞和成骨细胞。来自动物模型和 MRONJ 患者的数据显示 ARDs 对调节免疫细胞有深度干扰,尽管大部分文献涉及 BPs 的影响,但缺乏关于 Dmab 的数据,表明需要进一步研究。

pleurotus sajor-caju(Fr。)歌手β-1,3- ...
摘要:可食用的灰色牡蛎蘑菇,胸膜sajor-caju,β(1,3),(1,6)葡聚糖具有广泛的生物学活性,包括抗炎性,抗炎症,抗微生物和抗氧化剂。然而,其生物学活性受到高分子重量产生的低水溶性的限制。我们先前的研究表明,使用HEVEAβ-1,3-葡萄糖酶同工酶对灰色牡蛎蘑菇β-葡聚糖进行酶水解,可获得较低的分子量和较高的水溶性,Pleurotus sajor-sajor-caju-caju葡萄糖醇乙醇(PS-GOS)。此外,PS-GOS可能通过增强成骨细胞 - 骨形成来减少骨质疏松症,而其对骨细胞 - 骨的吸收的影响仍然未知。因此,我们的研究调查了PS-GOS在核因子Kappa-B配体(RANKL)诱导的骨化前肿瘤生成264.7细胞中核因子Kappa-B配体(RANKL)诱导的破骨细胞发生上的调节活性和潜在机制。PS-GOS在RAW 264.7细胞上的细胞细胞毒性由3-(4,5-二甲基噻唑-2-基)确定-2,5-二苯基-2H-2H-四唑溴化物(MTT)测定法,其对骨酸磷酸磷酸磷酸化酶(Trapsantase)(Trappase)的影响及其对骨质分化的影响。另外,通过坑形成测定,检测到其对破骨细胞骨敏感能力的影响。通过定量逆转录酶聚合酶链反应(QRT-PCR),Western blot和免疫流效来评估破骨细胞生成相关的因子。这些发现表明PS-GOS可能是作为骨代谢疾病的有效天然剂而有益的。结果表明,PS-GOS是无毒的,并有明显地抑制成熟破骨细胞多核细胞的形成及其吸收活性,通过减少诱捕阳性细胞的数量和PIT形成区域的数量,以剂量依赖性方式。此外,PS-GOS还减轻了活化B细胞的核因子Kappa轻链增强剂的核因子p65(NFκB-P65)的表达及其随后的主骨细胞调节剂,包括活化的T细胞C1(NFATC1)的核因子和FOS Proto proto proto-cogen-(CFOS)通过NF-NF-κB-B-B-κB-B b b b b b b b b。此外,PS-GOS明显抑制了等级表达,它是许多与破骨构成相关的级联反应的初始发射器,并抑制了蛋白水解酶,包括TRAP,基质金属肽酶9(MMP-9)和Cathepsin K(CTK)。

放射性核素刺激动态疗法正交靶向破骨细胞和骨髓瘤细胞诱导多维细胞死亡途径
原理:多发性骨髓瘤 (MM) 是一种骨髓浆细胞多灶性恶性肿瘤,其特征是缓解和复发的恶性循环,最终导致死亡。由于骨微环境 (BME) 和 MM 细胞 (MMC) 之间复杂的相互作用,该疾病大多无法治愈。在骨病的“恶性循环”中,MMC 对破骨细胞 (OC) 的异常激活会导致严重的骨溶解、促进免疫逃避并刺激 MMC 的生长。破坏这些癌症-基质相互作用将增强治疗反应。方法:为了打破这种循环,我们将载有非治疗剂量光敏剂二茂钛 (TC) 的纳米胶束 (NM) 正交靶向表达 VLA-4 (α 4β1、CD49d/CD29) 的 MMC (MM1.S) 和表达 α vβ3 (CD51/CD61) 的 OC。同时,全身施用非致死剂量的放射性药物 18 F-氟脱氧葡萄糖 ([ 18 F]FDG) 与 TC (放射性核素刺激疗法,RaST) 相互作用产生细胞毒性活性氧 (ROS)。在 MM1.S 细胞系以及异种移植和同种移植 MM 动物模型中表征了 RaST 的体外和体内作用。结果:我们的数据显示,RaST 诱导细胞脂质的非酶氢过氧化,最终导致线粒体功能障碍、DNA 碎片化和 MMC 的 caspase 依赖性凋亡,使用 VLA-4 亲和 TC-NMs。RaST 上调了 BAX、Bcl-2 和 p53 的表达,突出了通过 BAK 非依赖性途径诱导细胞凋亡。多铜氧化酶 F5 表达的增强(可抑制脂质氢过氧化和 Fenton 反应)不足以克服 RaST 诱导的不可逆功能扰乱 α,β-醛积累增加,这些醛会对 DNA 和蛋白质造成严重且持久的损害。在体内,VLA-4-TC-NM 或 α vβ3-TC-NMs RaST 均对免疫功能低下但免疫功能不正常的 MM 携带小鼠模型产生严重治疗效果。VLA-4-TC-NM 和 α vβ3-TC-NMs 联合治疗可协同抑制骨溶解、减轻肿瘤负担并防止两种 MM 体内模型中的快速复发。结论:通过同时靶向 MM 和骨细胞,联合 RaST 通过对骨癌恶性循环的多管齐下的作用抑制 MM 疾病进展。我们的工作没有采用标准的多药疗法,而是揭示了一种独特的光物理治疗模式,即使用无毒剂量的单一光敏药物正交地作用于癌症和骨细胞,然后通过放射性核素刺激产生 ROS 来抑制肿瘤进展并最大限度地减少免疫功能正常的小鼠和免疫功能低下的人类 MM 模型中的骨溶解。

stem-cell-therapy-brings new-life to-musculoskeletal- ...
再生电位PBSC是一种干细胞 - 一种基本和非专业细胞,具有开发(或分化)为其他类型细胞(例如骨骼和软骨)的潜力。PBSC已被证明具有产生间充质干细胞的潜力,间充质干细胞与肌肉骨骼再生特别相关,因为它们已经被启动以区分软骨细胞(软骨细胞)和骨细胞(骨细胞)(成骨细胞)。它们的再生潜力,结合了轻松收获和处理它们的能力,使PBSC成为有助于肌肉骨骼再生的出色候选者。更重要的是,可以在单个程序中大量收获PBSC,然后安全地存储几年,从而在初次治疗后数月后重复治疗。

短暂的研究沟通间充质干细胞在缺氧和地塞米松的预处理中促进骨细胞分化,在应激状态下
能够区分成骨细胞的骨髓衍生的间充质干细胞(MSC)用于有效再生疗法。必须提示MSC分化为成骨细胞,以使MSC移植有效。在这项研究中,评估了参与骨形成的成骨细胞分化标志物,以研究骨髓衍生的大鼠MSC对地塞米松和缺氧的应激抗性及其分化为骨细胞的能力。在三种不同的环境(地塞米松治疗,低氧条件[1%氧]或两者)中,允许MSC分化为成骨细胞21天。根据碱性磷酸酶水平和矿化测定法评估成骨细胞分化潜力。 免疫荧光染色用于确定成骨细胞分化标记I型胶原蛋白和骨桥蛋白的蛋白质表达。 MSC在缺氧条件下分化为成骨细胞,但在用来塞米松和地塞米松加上与对照相比缺氧后,分化的速度更慢。 MSC用地塞米松或缺氧预处理,然后允许在相似的条件下区分成骨细胞,从而与对照MSC相似。 MSC与不相比,对地塞米松或缺氧的抵抗力更快地分化为成骨细胞。 这些发现表明,通过地塞米松或缺氧暴露对MSC进行压力的阻力增加可能会导致移植后更快地分化为成骨细胞。成骨细胞分化潜力。免疫荧光染色用于确定成骨细胞分化标记I型胶原蛋白和骨桥蛋白的蛋白质表达。MSC在缺氧条件下分化为成骨细胞,但在用来塞米松和地塞米松加上与对照相比缺氧后,分化的速度更慢。MSC用地塞米松或缺氧预处理,然后允许在相似的条件下区分成骨细胞,从而与对照MSC相似。MSC与不相比,对地塞米松或缺氧的抵抗力更快地分化为成骨细胞。这些发现表明,通过地塞米松或缺氧暴露对MSC进行压力的阻力增加可能会导致移植后更快地分化为成骨细胞。

引用本文:Biswas L,Niveria K,Verma AK。活性氧在骨骼重塑中的悖论作用:骨质疏松和PO
骨质疏松症是一种代谢性骨病,它影响性别,并且是骨折最常见的原因。骨质疏松疗法主要抑制破骨细胞活性,很少旨在触发新的骨骼生长,从而经常引起严重的全身性不良反应。在生理上,细胞内氧化还原状态取决于促氧化剂,氧化剂(活性氧,ROS)和抗氧化剂的比率。ROS是骨质疏松症中氧化应激的关键因素,因为氧化还原状态的变化负责动态骨重塑和骨再生。ROS代和抗氧化剂系统中的失衡在骨质疏松症,刺激成骨细胞和骨细胞对破骨细胞生成的发病机理中起关键作用。ROS可防止矿化和成骨,从而导致骨质流失的增加。另外,抗氧化剂直接或间接地有助于激活成骨细胞,从而导致分化和矿化,从而减少骨质质外生的发生。由于免疫反应性的不可预测性和报告的不良反应,尽管药物对氧化应激产生了有希望的结果,但针对破骨细胞的临床治疗的治疗受到限制。纳米技术介导的干预措施比再生医学的其他治疗方式获得了显着的优势。纳米疗法方法通过增强其成骨和抗跨性栓塞潜力来影响纳米颗粒的抗氧化特性以触发骨骼修复,从而影响生物相容性,机械性能和骨诱导率。因此,利用纳米疗法来维持成骨细胞和破骨细胞的分化和增殖是典型的。

ROS 对牵张成骨过程中骨重塑的影响
细胞内氧化应激,特别是通过活性氧 (ROS),在牵张成骨 (DO) 过程中的骨骼重塑中起着关键作用,DO 是一种广泛用于骨骼修复和再生的骨科技术。本研究旨在阐明 ROS 在促进骨形成和骨吸收方面的双重作用,重点研究其对成骨细胞和破骨细胞活动的影响。利用体外和体内模型,我们测量了 DO 不同阶段(潜伏期、牵张和巩固)的 ROS 水平,并分析了它们对细胞功能和信号通路的影响。结果表明,牵张阶段的中等 ROS 水平可增强成骨细胞分化和骨矿化,而过度的氧化应激则促进破骨细胞活动和骨吸收。组织学和生化分析表明,ROS 不仅影响 Wnt/β-catenin 和 NF- κB 通路,而且还与炎症和血管生成过程相互作用,进一步影响骨愈合结果。这些发现强调了维持最佳 ROS 平衡以最大程度提高治疗效果和减少 DO 并发症的重要性。此外,该研究还强调了抗氧化剂疗法调节 ROS 水平的潜力,为改善骨再生的临床结果提供了新策略。这项研究弥补了对骨生物学氧化应激理解的关键空白,并为有针对性的干预措施以增强骨骼愈合铺平了道路。