XiaoMi-AI文件搜索系统
World File Search SystemAbeparvovec

主题:Onasemnogene Abeparvovec(Zolgensma)
第一种FDA批准的SMA治疗疗法是反义寡核苷酸的Nusinersen(Sprinraza)。它每4个月被用作鞘内输注。Zolgensma(Onasengene Abeparvovec-XIOI)是一种基于腺相关病毒载体的基因疗法,于2019年5月被批准用于治疗SMN1基因中具有双重性突变的SMA年龄小于2岁的儿科患者。它是通过诱导运动神经元和周围组织中SMN表达的单个一次性输注来给药。尚未评估重复给药的安全性和功效,也没有用于晚期SMA患者(例如,四肢完全麻痹,永久性呼吸机依赖性)。通过基因检测证实的1型SMA的个体(n = 15)证实的1型患者的1阶段开放标签研究(NCT02122952)评估了Zolgensma的安全性和功效。如果参与者年龄少于9个月,则包括参与者,有2份SMN2副本,并在6个月之前出现了症状(通过临床评估,运动技能延迟,头部控制效果不佳,头部控制效果不佳,肩部姿势和关节的过度运动)。那些需要侵入性呼吸机支撑或可以在不创侵入性呼吸机支持(BIPAP)上进行不到16个小时的人被排除在外。参与者根据Zogensma的剂量入学了两个人群。队列1(低剂量,n = 3)接受了6.7 x 10 13 vg/kg剂量,而队列2(高剂量,n = 12)接受了2.0 x 10 14 Vg/kg剂量。

对脊髓肌萎缩症患者体重≤17kg且≤24个月大的患者的骨nasegene abeparvovec的安全性和耐受性:
关键纳入标准•基于基因突变分析的症状SMA诊断,双重SMN1突变(缺失或点突变)和任何数量SMN2基因的副本•治疗时的年龄≤24个月•重量≤17kg筛选时访问时访问或批准的药物均可进行批准的药物,•纽约批准的药物/牢固的纽约,键/牢记的键abeparvovec在筛查前的四个星期内使用或先前使用任何AAV9基因疗法•抽吸史或吸气迹象(例如,食物的咳嗽或溅射)在筛查前的四个星期内在筛选前15天内

促进基因疗法的表达:更多并不总是更好
脊柱肌肉萎缩(SMA)是由SMN1的功能丧失引起的自身隐性神经肌肉疾病。SMA的特征是脊髓中运动神经元的变性,导致肌肉无力和萎缩。当前可用的三种可用治疗方法之一是基于AAV9的基因替代疗法的Abeparvovec。尽管它在改善SMA患者的运动功能方面有效,但其长期安全性仍然不清楚,诸如肝脏毒性等不良事件很常见。这可能是由高矢量剂量或超级生理水平的SMN引起的,这是由于其强,无处不在的启动子驱动的。在本期EMBO分子医学问题中,Xie等人通过内生SMN1启动子代替基准病毒的启动子(等效于Ona-emnogene Abeparvovec)来解决这一问题。在常见的SMA小鼠模型中,使用该第二代载体的治疗恢复了跨组织的生理水平的SMN表达,从而提高了安全性和有效性。这种方法对SMA和其他疾病的更安全,更有效的AAV基因疗法有希望。

包装传单:用户zolgensma®2×...
该药物受到其他监测。这将允许快速识别新的安全信息。您可以通过报告孩子可能会得到任何副作用来提供帮助。有关如何报告副作用,请参见第4节的结尾。在给您的孩子得到这种药物之前,请仔细阅读所有这些传单,因为它包含重要信息。- 保留此传单。您可能需要再次阅读。- 如果您还有其他问题,请询问孩子的医生或护士。- 如果您的孩子有任何副作用,请与孩子的医生或护士交谈。这包括此传单中未列出的任何可能的副作用。请参阅第4节。此传单中的内容1。什么是Zolgensma,以及它用于2。在给孩子Zolgensma 3.如何给出Zolgensma 4。可能的副作用5。如何存储Zolgensma 6。包装和其他信息的内容1。Zolgensma是什么以及用于Zolgensma的Zolgensma是一种称为“基因疗法”的药物。它包含活性物质onasengenogene abeparvovec,其中包含人类遗传物质。Zolgensma用于Zolgensma的方法用于治疗脊柱肌肉萎缩(SMA),这是一种罕见的,严重的遗传性疾病。ZOLGENSMA的工作方式SMA发生时,当需要制作一种称为“生存运动神经元”(SMN)蛋白质的必需蛋白质的基因中缺失或异常。缺乏SMN蛋白会导致控制肌肉(运动神经元)死亡的神经。2。这会导致肌肉变得虚弱并浪费,最终失去运动。该药物通过提供SMN基因的功能齐全的副本来帮助人体产生足够的SMN蛋白。使用不引起人类疾病的改良病毒,将基因传递到需要的细胞中。您需要在给孩子Zolgensma之前需要知道的内容不使用Zolgensma•如果您的孩子过敏于Onasemnogene Abeparvovec或该药物的其他任何成分(第6节中列出)。

Nusinersen(Spinraza)
•神经科医生的处方•患者对5q-独生体隐性隐性脊柱肌肉萎缩(SMA)的诊断为SMN1基因中的双重缺失或突变•患者患者具有2至4份SMN2基因的副本•患者没有使用SMA治疗的治疗方法。 (ZOLGENSMA)或其他用于SMA的基因疗法•患者可以无永久的入侵通气或气管造口术呼吸*•患者每天醒来时可以呼吸而无需入侵或非侵入性通风,在每天醒来期间,患者•KAISER永久性咨询的持续咨询及其在使用中,并在进行持续的医疗疗法(继续使用)。治疗期间每12个月进行一次审查)。在以下情况下建议中断:

NSD602-005.05
两种药物均已批准遵守PAS协议。鉴于这两种药物均已获得SMC批准并符合更新的UO定义,因此公元前将两种产品都包括在UO风险共享安排中。onasengene abeparvovec(Zolgensma)用于治疗SMA患者的治疗。基因疗法是基于成本的,并确保用于SMA的高成本药物的资金流量。risdiplam(evrysdi)用于治疗SMA的 SMC在2022年接受使用。 该药物是以成本为基础的,以确保用于SMA的高COS药物的公平资金流。 avallucosidase alfa(Nexviadyme)用于治疗庞贝疾病,SMC在2023年批准。 该药物已被添加到IMD风险共享中,因为它是药物藻糖苷酶AlfA(肌酶)的治疗替代品,该抗葡萄糖酶α(肌酶)已经分享了风险。SMC在2022年接受使用。该药物是以成本为基础的,以确保用于SMA的高COS药物的公平资金流。avallucosidase alfa(Nexviadyme)用于治疗庞贝疾病,SMC在2023年批准。该药物已被添加到IMD风险共享中,因为它是药物藻糖苷酶AlfA(肌酶)的治疗替代品,该抗葡萄糖酶α(肌酶)已经分享了风险。

HASLINK问题129
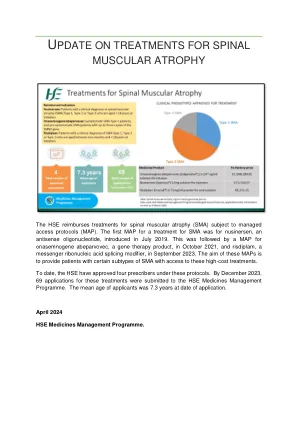
“每隔几个月必须去医院注射一次是一种创伤经历。一次性基因疗法的输注只需要一个小时。它可以消除长期治疗的不便,并给患者和全家带来新的希望。” HA首席药剂师Benjamin Lee博士解释说。SMA患者由于遗传缺陷而无法产生足够的功能性蛋白质,从而导致运动肌肉功能的丧失。新的基因疗法,即nasemenogene abeparvovec,涉及将相应基因携带到患者体内的无害病毒载体注入无害的病毒载体,这使基因可以执行其基本功能并补偿先天性缺陷,以便患者可以正常维持其肌肉功能,并避免使用vistilator。进行临床数据表明,新药的影响持续了长达7.5年后。

与SMA的症状前婴儿-ICNMD 2024
oa,不存在onsonem; SMA,萎缩肌肉脊柱。1。ma和al。在Neurol 2017:81:355-68中; 2。朋友A和Al。orphanes j Rare 2011; 6:71; 3。Govoni A和Al。 Neurobiol 2018; 55:6307–18; 4。 tw tw,最障碍的临床北部AM 2010; 37:23-36; 5。 Glascock J和Al。 J夜晚; 5:145–58; 6。 newson aj和al。 AJGP 2022; 51(3):131–5; 7。 澳大利亚的教会治理和老年护理。 %20cistic%20firosis,%20mouscular%20ATROPPHY%20fragille%20syndrome.pdf。 访问02 Octoo,2024。Govoni A和Al。Neurobiol 2018; 55:6307–18; 4。tw tw,最障碍的临床北部AM 2010; 37:23-36; 5。Glascock J和Al。J夜晚; 5:145–58; 6。newson aj和al。AJGP 2022; 51(3):131–5; 7。澳大利亚的教会治理和老年护理。%20cistic%20firosis,%20mouscular%20ATROPPHY%20fragille%20syndrome.pdf。访问02 Octoo,2024。

Swapnil Tirmanwar,Int。 J. of Pharm。 Sci。,2025,第3卷,问题01,131-136
脊柱肌肉萎缩(SMA)是一种主要影响运动神经元的遗传神经肌肉疾病,导致渐进的肌肉无力和萎缩。它是由SMN1基因突变引起的,这导致生存运动神经元(SMN)蛋白的水平降低,这对于肌肉功能必不可少。SMA以不同类型的形式呈现,从严重的婴儿发作形式到较轻的成人发作病例,以及影响流动性,呼吸和运动发育的症状。此临床评论概述了SMA的遗传基础,分类,症状,症状,症状和诊断方法。它还研究了治疗策略的进步,包括基因疗法(例如Onasemnogene Abeparvovec),SMN2剪接修饰剂(如Nusinersen)以及支持护理以改善生活质量。该评论进一步强调了早期诊断,疗法的可及性以及新生儿筛查计划对更好结果的重要性。目标是对SMA进行全面的了解,以告知临床医生,看护人和公共卫生从业人员有效的疾病管理和新兴治疗选择。