XiaoMi-AI文件搜索系统
World File Search SystemErlotinib

版权材料
在癌症治疗中,最初的开发努力是通过寻找对癌细胞系有毒性的药物来经验性地发现癌症治疗方法。随着对癌症生物学的深入了解,人们确定了特定的癌症靶点,例如激素受体 (HR)(雌激素、孕酮、雄激素)、人类表皮生长因子家族受体 (HER2、EGFR)、血管内皮生长因子 (VEGF) 受体等。表皮生长因子受体 (EGFR) 靶向治疗的发展历史可能最能体现如何最大限度发挥靶向治疗效果的挑战。最初,EGFR 靶向药物,如吉非替尼和厄洛替尼,被用于治疗所有非小细胞肺癌 (NSCLC) 患者,但只有一小部分患者受益于该疗法。几年后,人们意识到只有患有 EGFR 蛋白突变的肺癌肿瘤的人才会有显著的反应。

EGFR 靶向治疗的肾脏安全性
简单总结:针对上皮生长因子受体 (EGFR) 的药物用于治疗肺癌和消化道癌症,代表着医学的重大进步。除了定位于癌细胞外,EGFR 还可在肾脏中找到。这一观察结果引发了这些药物肾毒性的问题。本研究基于来自最大的国际药物警戒数据库 VigiBase ® 的安全数据进行了研究,解决了这个问题。本研究表明,这些药物的肾毒性主要表现为消化毒性下的肾衰竭。厄洛替尼是首个获得市场授权的抗 EGFR 药物,已发现一种称为溶血和尿毒症综合征或血栓性微血管病的新不良反应。这个信号必须得到确认。尚未发现与抗 EGFR 药物相关的其他肾毒性,特别是肾小球毒性或肾小管毒性。
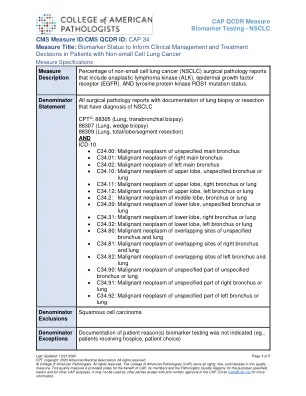
CAP 34 措施标题:生物标志物状态用于指导临床...
这一重要信息对于协调护理和有效利用资源至关重要。因此,了解 ROS1、ALK 和 EGFR 的突变状态对于晚期 NSCLC 的适当临床决策是必不可少的。如果在一线化疗之前或期间发现一种或多种这些重排,则应考虑替代疗法。1% 至 2% 的非小细胞肺癌会出现 ROS1 重排,并且可预测对一线治疗药物克唑替尼和色瑞替尼疗法的反应。反应率(包括完全反应)接近 70% (2-4)。对于 ALK1,除了识别可能对靶向疗法有反应的肿瘤外,了解 ALK 重排状态通常可预测对免疫疗法的反应较差。大约 5% 的肺腺癌具有涉及 ALK 基因的染色体重排,并与 ALK 蛋白过表达有关。患有此类肿瘤的患者对 ALK 酪氨酸激酶抑制剂(如克唑替尼)的治疗有反应 (2, 5)。最后,约 20% 的肺腺癌含有 EGFR 激活突变,这预示着对 EGFR 酪氨酸激酶抑制剂(如厄洛替尼)的治疗有反应 (2, 6-10)。

伏立诺他通过上调 PTEN 诱导乳腺癌细胞 G2/M 细胞周期停滞
摘要。– 目的:乳腺癌 (BC) 是全世界女性中最常见的癌症类型。人们提出了各种方法来治疗这种疾病,但没有一种单一的药物被证明是有效的。因此,了解不同药物的分子机制变得势在必行。本研究旨在评估厄洛替尼 (ERL) 和伏立诺他 (SAHA) 在诱导乳腺癌细胞凋亡中的作用。还根据一些癌症相关基因的表达谱评估了这些药物的作用;PTEN、P21、TGF 和 CDH1。材料和方法:在本研究中,乳腺癌细胞 (MCF-7) 和 MDA-MB-231 以及人羊膜细胞 (WISH) 用两种浓度 (50 和 100 µM) 的厄洛替尼 (ERL) 和伏立诺他 (又名 SAHA) 处理 24 小时。收获细胞用于下游分析。通过流式细胞仪分析DNA含量和细胞凋亡,并进行qPCR以评估不同癌症相关基因的表达。结果:结果表明,与正常细胞和对照相比,ERL和SAHA在24小时后将两种乳腺癌细胞都抑制在G2/M期。对于细胞凋亡,随着两种所用药物的浓度增加,BC细胞显示出升高的总细胞凋亡水平(早期和晚期),在24小时处理中,ERL的最有效浓度为100µM。在对照细胞中,SAHA被证明是在100µM浓度下最有效的药物,在24小时处理中细胞凋亡百分比范围为1.7-12%。在所使用的两种乳腺癌细胞系中,坏死也呈剂量依赖性。我们进一步评估了 PTEN 、 P21 、 TGF- β 和 CDH1 的表达谱。在 MCF-7 中,数据表明对于 TGF- β 、 PTEN 和 P21 ,最有效的治疗是浓度为 100 µM 的 SAHA,而对于 CDH1 ,最有效的浓度是 100 µM 的 ERL。在 MDA-MB-232 中也观察到了类似的情况,其中对于 TGF- β 、 PTEN 和 P21 ,最有效的治疗是浓度为 100 µM 的 SAHA,而对于 CDH1 ,最有效的浓度是 50 µM 的 SAHA。结论:我们的结果揭示了 ERL 和 SAHA 在调节癌症相关基因表达中的作用,尽管这些数据需要进一步研究。

外推随机抽取等离子体样品到谷水平的可行性,用于治疗药物监测的小分子激酶抑制剂
摘要:小分子激酶抑制剂(SMKI)广泛用于肿瘤学。用于SMKI的治疗药物监测(TDM)可能会减少不受欢迎或过度暴露。但是,诸如戒断时间的后勤问题阻碍了其在临床实践中的实施。使用消除半衰期将随机浓度推送到槽浓度可能是克服这个问题的一种简单简单的方法。在我们的研究中,在24小时采样期间观察到的血浆浓度用于外推到谷水平。目的是证明与测得的c min值相比,随机抽血样品的外推将导致等效估计的谷物样品。总共分析了2241个血液样本。如果在t最大后绘制样品,则估计的afatinib和sunitinib的水平满足了等效标准。在药物摄入后,erlotinib,伊马替尼和索拉非尼的计算c槽水平分别符合等效性迹象。对于再丙替尼的外推不可行。总而言之,使用平均消除半衰期将随机服用的药物浓度外推到谷浓度,对于多个SMKI是可行的。因此,这种简单的方法可以积极地促进TDM在肿瘤学中的实现。

通过上调葡萄糖-6- ...
胰腺导管腺癌(PDAC)是最恶性的癌症形式之一。缺乏有效的治疗选择和耐药性会导致PDAC患者的生存率较低。在这项研究中,我们研究了对EGFR抑制剂Erlotinib不反应的胰腺癌细胞的代谢改变。我们从MIAPACA2和ASPC1细胞系中选择了抗洛替尼的胰腺癌细胞。与敏感细胞相比,埃洛替尼抗性细胞的代谢分析表明,糖酵解活性的显着下调和糖酵解代谢物的水平降低。抗性细胞表现出参与ROS调节和核苷酸生物合成的五磷酸途径(PPP)酶的表达升高。增强的PPP升高的细胞NADPH/ NADP+比率,并保护细胞免受活性氧(ROS)诱导的损伤。使用6-氨基氨氨酰胺(6AN)升高ROS水平,诱导G1细胞周期停滞,并将抗性细胞抑制PPP。遗传研究确定升高的PPP葡萄糖-6-磷酸脱氢酶(G6PD)是促进性厄洛替尼耐药的重要促进者。从机械上讲,我们的数据表明,分化抑制剂(ID1)的上调调节抗性细胞中的G6PD表达,从而导致代谢表型改变并减少对厄洛替尼的反应。一起,我们的结果突出了肿瘤代谢在PDAC药物反应中的潜在作用,并识别G6PD是克服耐药性的靶标。

第四代表皮生长因子受体...
非小细胞肺癌(NSCLC)中表皮生长因子受体(EGFR)突变是最常见的驱动突变之一,尤其在某些人群中,例如亚洲患者和非吸烟者。外显子19的缺失和外显子21的L858R点突变是最常见的异常,两者合计占所有EGFR突变的80%以上(1)。在过去的二十年里,靶向治疗的出现深刻改变了晚期驱动基因阳性NSCLC患者的治疗策略。作为NSCLC的重要治疗靶点,EGFR酪氨酸激酶抑制剂(EGFR-TKI)彻底改写了EGFR突变型NSCLC患者的诊断和治疗(2)。与传统化疗相比,第一代(如吉非替尼和厄洛替尼)和第二代(如阿法替尼)EGFR-TKI已显示出更高的反应率和无进展生存期(PFS)。第三代 EGFR-TKI(如奥希替尼)的开发主要是为了克服由于 T790M 耐药突变而对早期 EGFR-TKI 产生的获得性耐药性,这是对早期 EGFR-TKI 产生耐药性的常见机制 (3)。奥希替尼还显示出作为 EGFR 突变 NSCLC 患者的一线治疗药物的疗效,因为它能够靶向常见的激活性 EGFR 突变和 T790M

基于纳米颗粒的晚期胰腺癌药物输送...
胰腺导管腺癌 (PDAC) 在所有疾病阶段的 5 年总生存率 (OS) 都很低,自 2012 年以来仅增加到 12%。对于出现转移性 PDAC 的患者,这一比例下降到 3% (1)。根据全球癌症观察站 (GLOBOCAN) 2020 年的数据,PDAC 每年导致超过 466,003 人死亡 (2)。基因组研究工作使人们对突变和结构格局有了更深入的了解,以致癌突变为主,90% 的患者被确诊为 Kirsten 大鼠肉瘤病毒致癌基因 (KRAS) 突变。然而,尽管最近在支持 KRAS 抑制剂治疗 PDAC 方面取得了进展,但可能会产生耐药性,化疗将继续成为 PDAC 管理的支柱。这强调了迫切需要找到更好的治疗方案并增强药物输送机制。PDAC 联合治疗的首批阳性 III 期试验之一是加拿大癌症试验组 (CCTG) PA.3 将表皮生长因子受体 (EGFR) 抑制剂厄洛替尼与吉西他滨 (3) 相结合。然而,由于缺乏生物标志物来丰富反应,因此临床上并不认为这种益处相关。从那时起,更多的组合已成为标准实践。

胰腺癌的靶向治疗
没有动脉和/或有限的静脉与血管接触,只有 10-15% 的患者符合这些标准 (5)。对于患有转移性晚期疾病或 PDAC 复发的患者,细胞毒性化疗方案是标准治疗,总生存期 (OS) 在数周至数月之间 (4)。单药吉西他滨于 1997 年获批,尽管临床反应不佳,中位生存期约为 6 个月 (6),它仍然是 PDAC 的标准治疗方法超过二十年。表皮生长因子受体 (EGFR) 抑制剂厄洛替尼与吉西他滨联合使用,与单独使用吉西他滨相比,PDAC 患者的 OS 延长了 10 天,并于 2005 年获得 FDA 批准 (7)。 2011 年,一种更强烈的化疗方案 FOLFIRINOX(奥沙利铂、伊立替康和氟尿嘧啶/亚叶酸钙)获批用于 PDAC 治疗,生存期延长约 11 个月 (8)。然而,正如预期的那样,这种方案的毒性更高,因此只有体能状态较高的患者才有资格接受这种治疗。2013 年,白蛋白紫杉醇(一种白蛋白结合的紫杉醇制剂)与吉西他滨 (NPT + Gem) 联合使用,中位生存期为 8.5 个月,这促使 FDA 批准这种组合作为 PDAC 患者的一线治疗方案 (9)(表 1)。

抑制 CDK8/19 介导激酶可防止对 EGFR 靶向药物产生耐药性
摘要:耐药性是实现常规和靶向抗癌药物治愈的主要障碍。获得性耐药性的出现最初是由非遗传转录变化介导的,这种变化发生的频率比突变高得多,可能涉及群体规模的转录组适应。CDK8/19 激酶通过与转录介导复合物结合,与不同的信号响应转录因子协同调节转录重编程。在这里,我们测试了 CDK8/19 抑制是否可以阻止对作用于表皮生长因子受体 (EGFR/ERBB1/HER1) 的药物的适应。在 BT474 和 SKBR3 乳腺癌细胞长期暴露于 EGFR 靶向小分子 (吉菲替尼、厄洛替尼) 以及 SW48 结肠癌细胞长期暴露于抗 EGFR 单克隆抗体西妥昔单抗后,分析了耐药性的发展。在所有情况下,用单剂量药物治疗小细胞群(~10 5 个细胞)最初会导致生长抑制,随后适应群体中增殖恢复并产生耐药性。然而,这种适应总是通过添加选择性 CDK8/19 抑制剂来阻止,即使此类抑制剂单独对细胞生长只有中等或没有影响。这些结果表明,将 EGFR 靶向药物与 CDK8/19 抑制剂相结合可能会延迟或阻止肿瘤对治疗产生耐药性。