XiaoMi-AI文件搜索系统
World File Search SystemG12C

对RAS抑制剂和癌症免疫疗法的事实和希望Jesse Boumelha,Miriam Molina-Arcas和Julian Downward
摘要◥尽管过去十年在免疫疗法的开发中取得了长足的进步,从而使免疫系统反对肿瘤,但在阻断癌症生长的致癌驱动因素的药物方面也取得了重大进展。但是,将免疫性与靶向致癌驱动器途径的药物结合在一起的进展很少。人类癌症中一些最重要的癌基质编码了RAS家族蛋白,尽管这些蛋白质被证明对靶标有挑战。最近已批准抑制KRAS:G12C的特定突变体形式的药物。这些已改善了具有这种突变的肺癌患者的治疗,但是初始反应后获得的耐药性耐药性的发展限制了对整体生存的影响。由于免疫抑制
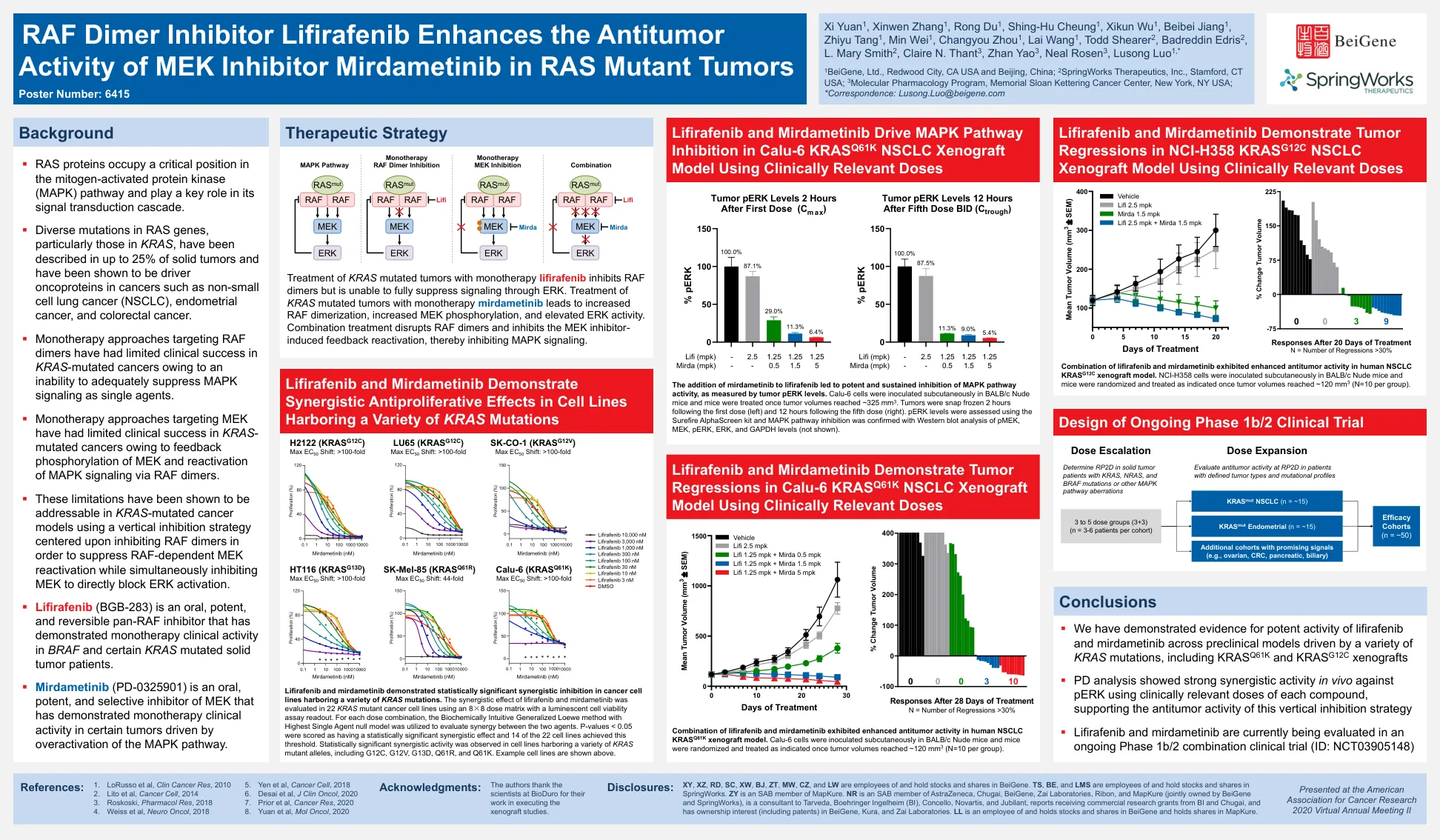
RAF二聚体抑制剂lifirafenib增强了MEK抑制剂米尔德梅尼在RAS突变肿瘤中的抗肿瘤活性
lifirafenib和mirdametinib在具有多种KRAS突变的癌细胞系中表现出具有统计学意义的协同抑制作用。使用8×8剂量基质在22个KRAS突变癌细胞系中评估了Lifirafenib和Mirdametinib的协同作用,并具有发光细胞活力测定能力读数。对于每种剂量组合,使用具有最高单药物无效模型的生化直觉的概括性LOEWE方法来评估两种药物之间的协同作用。p值<0.05被评分为具有统计学意义的协同作用,而22个细胞系中有14个达到了该阈值。在具有各种KRAS突变等位基因的细胞系中观察到了统计学上显着的协同活性,包括G12C,G12V,G13D,Q61R和Q61K。示例细胞系如上所述。

回顾一种姗姗来迟的针对非小细胞肺癌 KRAS 突变的靶向治疗:聚焦 Adagrasib
直到最近,尽管 KRAS 突变是最常见的致癌驱动因素之一,但其在实体瘤治疗中仍未得到满足。从历史上看,KRAS 突变很难靶向,这是因为其整体表面结构光滑,缺乏结合位点,尺寸小,且对 GTP/GDP 的亲和力极高。21、22 2013 年,Ostrem 等人首次发现了靶向 KRAS G12C 的潜力,他们发现结合变构口袋可以将 GDP 结合的 KRAS 锁定在其非活性状态。23 研究表明,RAS 蛋白第 12 或 13 个密码子的突变会损害 GTP 水解,使 RAS 处于 GTP 结合的活性状态。24 这导致了 sotorasib 的开发,这是同类中首批 KRASG12C 失活状态抑制剂之一,并最终促使 FDA 批准 sotorasib 用于治疗

KRAS、BRAF、BRCA1 和 EGFR 突变特异性面板...
针对突变的疗法已成为一种变革性技术,在许多方面,它已成为癌症患者治疗的新范式。早期开发用于治疗非小细胞肺癌的厄洛替尼为 EGFR 治疗铺平了道路,无论肿瘤适应症亚型如何。最近推出的用于治疗 KRAS G12C 突变的 Sotorasib 标志着曾经无法用药的突变的转折点。基因特异性治疗的出现需要重新配置临床前模型,以更有效地代表不同背景和肿瘤适应症人群中的上述突变。3D 模型能够重现癌症的主要特征 - 例如生长、侵袭、免疫浸润和抑制、基质转化、药物扩散以及基因突变特征 - 对于推动快速和可扩展的化合物功效筛选具有重要意义。

krazati,inn-adagrasib
该药物会受到其他监测。这将允许快速识别新的安全信息。医疗保健专业人员被要求报告任何可疑的不良反应。有关如何报告不良反应的第4.8节。1。药用产品的名称Krazati 200 mg薄膜涂层片2。定性和定量组成,每个薄膜涂层的片剂包含200毫克Adagrasib。有关赋形剂的完整列表,请参见第6.1节。3。制药形式胶片涂层的片剂。白色至灰白色的椭圆形,薄膜涂层的平板电脑,约8 x 16毫米,一侧有风格化的“ M”,另一侧标有“ 200”。4。临床细节4.1治疗指示Krazati作为单一疗法,用于治疗成年患者,即至少一种先前的全身治疗后,具有KRAS G12C突变和疾病进展的晚期非小细胞肺癌(NSCLC)。4.2 krazati的生态学治疗方法应由抗癌药物使用的医生开始。必须使用经过验证的测试来确认KRAS G12C突变的存在,然后再开始使用Krazati进行治疗。posology建议的克拉萨蒂剂量为600毫克(三个200毫克片剂),每天两次。建议对Krazati进行治疗持续时间,直到疾病进展或不可接受的毒性为止。应告知患者延迟或错过的剂量患者,如果自预定给药时间以来少于4小时,则患者应正常服用剂量。如果错过了超过4个小时的剂量,则应跳过剂量,并且应在下一个预定剂量时恢复剂量。如果服用剂量后发生呕吐,则应建议患者不要服用额外的剂量。应按照规定服用下一个剂量。治疗期间的剂量调整建议的不良反应管理剂量降低水平在表1中概述了。

KRAS突变非小细胞肺癌 人与机器人的相互作用和神经经济学 kshv重编程宿主能量代谢用于发病机理 组合化学免疫疗法的疗效是针对HER2改变的晚期非小细胞肺癌患者的一线治疗:病例系列 IL-17抑制剂相关炎症性肠病 与可再生能源集成的电力系统的全生命周期管理:概念,发展和观点 框架对COVID-19疫苗抗性的影响 gompertz法律清洁泡沫合并 欧洲的能量和发射效率的演变... 基于低音模型的新能源行业中领先技术的扩散机制 从枯草芽孢杆菌增强的Menaquinone-7生产增强的进步 折面:中枢神经系统疾病的基因组编辑 质体生物学的基因组编辑 基因组编辑人多能干细胞,以建模β-... 改善了用不间断激发的双重喂养的风力涡轮机的短路电流计算 经济政策不确定性和金融创新 自然条件下的情感神经反馈 对绝热量子计算机上的循环量子重力模拟的前奏
肺癌是全球癌症相关死亡的主要原因,可以分为小细胞肺癌和非小细胞肺癌(NSCLC)。NSCLC是最常见的组织学类型,占所有肺癌的85%。NSCLC中常见的Kirsten大鼠肉瘤病毒癌基因(KRAS)突变与预后不良有关,这可能是由于对大多数全身疗法的反应不良,并且缺乏靶向药物。有关新的小分子KRAS G12C抑制剂,AMG510和MRTX849的最新发表的临床试验数据,表明这些分子可能有可能有助于治疗KRAS突变的NSCLC。同时,在免疫治疗过程中,在患有KRAS突变的患者中观察到了免疫效率。在本文中,综述了本文的发病机理,治疗状况,免疫疗法的进展以及KRAS突变NSCLC的靶向治疗。

靶向胰腺癌中的KRAS突变
针对RAS途径仍然是精确肿瘤学的圣杯。在胰腺导管腺癌(PDAC)的情况下,癌基因KRAS中的港口突变为90-92%,从而触发了规范的MAPK信号传导。过去没有结合口袋的KRAS蛋白质的平滑结构及其对GTP的亲和力在过去妨碍了药物的发展。KRAS G12C共价抑制剂的出现为瞄准KRAS提供了新的热情。然而,与RAS激活有关的众多途径确实导致了早期抗性的发展。此外,由致癌性KRAS决定的致密基质细分市场和免疫抑制微环境可能会影响治疗反应,从而强调了对基于组合的方法的需求。鉴于KRAS的突变发生在PDAC肿瘤发生早期,因此对其多效性作用的理解是该疾病进展的关键。在此,我们回顾了针对KRA的当前观点,重点是PDAC。

基于 PROTAC 的癌症联合疗法
背景 PROTAC(蛋白水解靶向嵌合体)代表了一类有前途的新型药物,可选择性地降解细胞中的目标蛋白质。PROTAC 是具有两个功能端的小分子,一个小分子端与目标蛋白质结合,另一端与 E3 泛素连接酶结合。PROTAC 成分将泛素连接酶募集到目标蛋白质,导致其泛素化并随后被蛋白酶体降解。PROTAC 已被开发用于多种癌症靶标,包括致癌激酶、表观遗传靶标和最近的 KRAS G12C 蛋白,其中几种目前正在临床试验中针对各种癌症进行测试。在临床前癌症模型中已报告对 PROTAC 的获得性耐药性,这表明 PROTAC 疗法对癌症的长期益处可能有限。因此,需要一种能够克服对 PROTAC 的耐药性并提供持久药物反应的治疗方法。发明概述

RMC- ...rtk信号传导和WT RAS活性是对GDP状态抗性的肿瘤的脆弱性
•Sotorasib(AMG-510)和Adagrasib(MRTX849)在具有致癌性KRAS G12C突变的肺癌患者中表现出临床益处。•初始反应的深度/速率以及单一疗法益处的耐用性都受到电阻发作的限制。•最近在临床前模型和临床样本中发布的数据确定了RAS同工型或其他旁路基因组病变中的其他致癌改变是获得性抗性的潜在机制。但是,与一半以上患者相关的耐药性的特征未通过可用的治疗样品的基因组测序来定义。•在这项研究中,通过多模式基因组,转录组和质谱法基于磷酸蛋白酶学的分析,对几种临床前的sotorasib和Adagrasib抗性模型进行了分析,我们将信号网络的重新透射视为肿瘤逃生的非基因组机制。•我们进一步确定了RAS(ON)多选择性抑制剂及其组合是克服对该机制抗性的治疗方法。

非小细胞肺癌的生物学见解
摘要肺癌的发生依赖于细胞内的半胱氨酸来克服氧化应激。包括非小细胞肺癌 (NSCLC) 在内的几种肿瘤类型通过过表达胱氨酸转运蛋白 SLC7A11 上调 xc - 胱氨酸/谷氨酸反向转运蛋白 (xCT) 系统,从而维持细胞内半胱氨酸水平以支持谷胱甘肽合成。核因子红细胞 2 相关因子 2 (NRF2) 通过调节 SLC7A11 充当氧化应激抵抗的主要调节器,而 Kelch 样 ECH 相关蛋白 (KEAP1) 充当氧化反应转录因子 NRF2 的细胞质抑制因子。KEAP1/NRF2 和 p53 的突变会诱导 NSCLC 中的 SLC7A11 激活。细胞外胱氨酸对于提供对抗氧化应激所需的细胞内半胱氨酸水平至关重要。胱氨酸可用性中断会导致铁依赖性脂质过氧化,从而导致一种称为铁死亡的细胞死亡。xCT 的药理抑制剂(SLC7A11 或 GPX4)会诱导 NSCLC 细胞和其他肿瘤类型的铁死亡。当胱氨酸摄取受损时,细胞内的半胱氨酸池可以通过转硫途径维持,该途径由胱硫醚-B-合酶 (CBS) 和胱硫醚 g-裂解酶 (CSE) 催化。外源性半胱氨酸/胱氨酸和转硫途径参与半胱氨酸池和下游代谢物会导致 CD8 + T 细胞功能受损和免疫疗法逃避,从而削弱免疫反应并可能降低免疫治疗干预的有效性。细胞焦亡是一种以前未被认识的受调节细胞死亡形式。在由 EGFR、ALK 或 KRAS 驱动的 NSCLC 中,选择性抑制剂可诱导细胞焦亡和凋亡。靶向治疗后,线粒体内在凋亡途径被激活,从而导致 caspase-3 的裂解和活化。因此,gasdermin E 被激活,从而导致细胞质膜通透化和细胞溶解性焦亡(以特征性细胞膜膨胀为标志)。本文还讨论了 KRAS G12C 等位基因特异性抑制剂的突破和潜在的耐药机制。关键词溶质载体家族 7 成员 11 (SLC7A11);核因子红细胞 2 相关因子 2 (NRF2);铁死亡;焦亡;KRAS G12C 等位基因特异性抑制剂;非小细胞肺癌 (NSCLC)