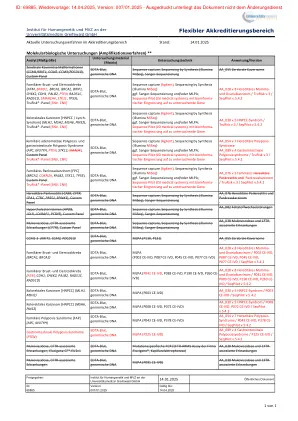
XiaoMi-AI文件搜索系统
World File Search SystemIllumina

RNA测序中基因表达模式的比较分析
使用Ampure XP珠和Magflo ngs珠的方法在整个Illumina Truseq RNA库制备工作流程(图1)中处理RNA样品(图1),然后在Illumina Novaseq(2 x 100 bp)上进行测序。随后,使用FASTQC工具进行了测序质量评估。使用火山图(图4)可视化差异基因表达分析,该图描绘了显着性与倍数变化值,从而可以鉴定2个条件之间基因表达的统计学意义变化。此外,代表前30个上调基因和下调基因的热图通过不同的基于珠子的纯化方法对整体表达模式提供了见解(图5)。通过Microsynth进行了实验程序和随后的生物信息学分析。

08:55 09:30 10:30 11:00
Emma Baple,埃克塞特大学基因组医学教授 Andrew Parrish,埃克塞特基因组学实验室系主任 Sean Humphray,Illumina 转化研究、应用与客户支持高级总监

aurolineatum(Haemulidae,lutjaniformes)的完整基因组序列,Tomtate Grunt
使用一个野外收集的标本进行测序。DNA提取。根据制造商的说明,使用Illumina Truseq套件构建了配对的测序库。该库是在配对端,2×150 bp格式的Illumina Hi-Seq平台上进行测序的。用三型V0.33(Bolger,Lohse和Usadel 2014)修剪了所得FASTQ文件的适配器/引物序列和低质量区域。修剪序列由黑桃v2.5组装(Bankevich,Nurk,Antipov等2012)随后使用Zanfona V1.0(Kieras 2021)进行完成步骤,以基于相关物种中保守的区域加入附加的重叠群。

补充材料
补充材料。材料与方法文库制备和 Miseq (Illumina®) 测序使用文库制备指南 (LPG) ( https://support.illumina.com/downloads/16s_metagenomic_sequencing_library_preparation.html ) 中报告的 Illumina 接头序列和引物悬垂部分(正向和反向)扩增 16S rRNA 基因的 460 bp V3-V4 高变区。使用以下 PCR 反应扩增每个 DNA 样本:2.5 µl 5 ng/ µl DNA、5 µl 引物正向悬垂部分、5 µl 引物反向悬垂部分、12.5 µl 2x KAPA HiFi HotStart ReadyMix (KAPA Biosystems)。使用 LPG 中报告的循环程序。 PCR 产物在 2% 琼脂糖凝胶(GellyPhor LE,Euroclone SPA,意大利米兰)上电泳分离,并用 GelRed™ 核酸凝胶染料(Biotium,美国加利福尼亚州海沃德)染色。通过紫外光透射仪观察预期长度的 PCR 产物的存在。然后,用 NucleoMag 试剂盒纯化 DNA 扩增子以清理和选择 NGS 文库制备反应的大小(Macherey-Nagel),并按照制造商的说明使用 Illumina® DNA/RNA UD Indexes Tagmentation 试剂盒对每个样本进行索引。在验证和定量之前,对文库进行进一步纯化。在 Agilent 4150 TapeStation D1000 ScreenTape 检测仪(安捷伦科技公司)上对文库进行验证,以验证大小,而定量则使用 Qubit 4 荧光计(赛默飞世尔科技,美国)。根据 DNA 扩增子的大小,应用 Illumina LPG 中报告的公式,以 nM 为单位计算最终的 DNA 浓度。最后,将每个文库中的 5 µl 稀释 DNA 等分试样混合,以合并具有唯一索引的文库。在 Miseq 加载之前,根据 Illumina LPG 说明对合并的文库进行变性和稀释。使用 MiSeq Reagent Micro Kit v2(500 个循环)加载合并的文库,运行包括 20% PhiX 作为内部对照。生物信息学分析测序数据包含在包含带有原始读取的 FASTQ 文件的文件夹中(R1 文件包含每个样本的正向读取,R2 文件包含每个样本的反向读取),使用 FastQC(英国剑桥 Babraham Institute)进行质量检查。然后,使用 DADA2 R 包(Callahan 等人,2016 年)处理 R1 和 R2 文件以生成扩增子序列变体 (ASV)(图 1)。最终生成了 ASV 表,总结了每个样本的不同 ASV 的数量。

氟康唑抗性的解脂耶氏酵母临床分离株 CBS 18115 的基因组序列草图
arrowia lipolytica 属于子囊菌门、酿酒菌亚门和双足菌科 (1)。除了工业用途 (2) 之外,Y. lipolytica 还广泛存在于食品、环境和动物中 (1)。由于其能够在 32°C 以上不稳定地生长,因此通常认为该菌种可安全用于工业用途 (1)。Yarrowia lipolytica 是一种机会性病原体,可引起侵袭性念珠菌病 (3)。在体外,该菌种被认为对氟康唑敏感 (4)。第一个 Y. lipolytica 基因组 (CLIB122) 于 2004 年发布 (5)。我们报告了对氟康唑有抗性的 Y. lipolytica 临床分离株的基因组草图,该分离株是从溃疡性结肠炎手术后的血培养中采集的。有趣的是,尽管之前曾接触过唑类药物,但使用梯度浓度试纸法(Etest;bioMérieux),该菌株的氟康唑 MIC 为 0.256 mg/mL。患者成功地用卡泊芬净治疗。该菌株在 35°C 的显色琼脂平板(CAN2;bioMérieux)上生长,并使用 Vitek 基质辅助激光解吸电离 - 飞行时间质谱 (MALDI-TOF MS) 仪器(bioMérieux)进行鉴定。在溶菌酶细胞壁消化后,使用 QIAmp DNA minikit(Qiagen)提取基因组 DNA。使用 Illumina DNA 制备标记试剂盒(Illumina)构建文库。简而言之,使用珠状转座子技术和集成 DNA 技术 (IDT) 的 Illumina DNA/RNA 独特双重 (UD) 索引集将 30 ng 总 DNA 片段化并进行索引。使用 Qubit 高灵敏度试剂盒 (Thermo Fisher Scienti ) 对文库进行扩增、纯化和定量。最后,将 9 pM 汇集和变性文库放入 2 250-bp v2 试剂盒 (Illumina) 中,并使用 MiSeq 仪器 (Illumina) 进行测序。使用 CLC Genomics Workbench v22.0 (Qiagen) 中的 Trim Reads v2.5 和 De Novo Assembly v1.5 工具对原始读取进行修剪、组装成重叠群并进行搭建。使用覆盖率与长度图丢弃覆盖率为 , 10 且长度为 , 500 bp 的重叠群 (6)。使用 QUAST v5.0.2 对最终的 scaffold 集进行质量分析 (7)。总基因组大小为 20,255,408 bp,分布在 521 个 scaffold 上(覆盖率为 100 ),N 50 值为 105 kbp(最长 scaffold,397 kbp),GC 含量为 49.03%。AUGUSTUS v3.4.0 (8) 使用白色念珠菌训练数据集预测了 6,151 个蛋白质编码基因,使用 tRNAscan-SE 2.0 检测到了 484 个 tRNA 基因 (9)。使用 BUSCO v5.3.2 和 saccharomycetes_odb10 谱系数据集 (10) 估计基因组完整性为 95.3%。平均核苷酸同一性 (ANI) 计算
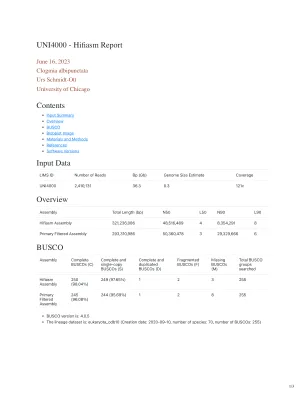
输入数据概述BUSCO UNI4000 -CDN
对于每个DiDail omni-c文库,将染色质与甲醛固定在原子核中,然后提取。用DNase I消化了固定的染色质,将染色质末端修复并连接到生物素化桥适配器,然后将含有末端的衔接子接近粘合。接近连接后,将交联后逆转并纯化了DNA。纯化的DNA以去除未结扎片段内部的生物素。使用NEBNEXT Ultra酶和Illumina兼容适配器生成测序文库。在每个文库富集之前,使用链霉亲和素珠分离含生物素的片段。库是在Illumina Hiseqx平台上测序的,以产生约30倍的序列覆盖率。然后Hirise使用MQ> 50读脚手架的读数(有关数字,请参见上面的“读取对”)。

使用人DNA样品识别试剂盒的说明
手册。a。我们建议长度为250 bp,以计算库摩尔力。b。所有目标从读取开始之初就位于50 bp之内,这意味着≥75bp的读数长度足以执行所有目标的分析。c。单源样本所需的读数最小的50倍覆盖范围为每个样本的总读数为7500。d。建议用于QC的5%phix尖峰。e。将图书馆池稀释到Illumina仪器所需的装载浓度。我们建议从初始序列运行的较低加载浓度开始,并在需要时进行调整。这避免了过度集群和运行的潜在失败。有关测序指南,请参见表1。表1 Illumina Sequencer和样品多路复用指南

人工智能道德原则
Illumina 致力于通过释放基因组的力量来改善人类健康。这一使命驱动着我们所做的一切,包括我们开发的技术。Illumina 创建并使用人工智能 (AI) 系统来提供行业领先的测序质量、推动数据洞察、提高对与健康和疾病相关的基因组变异的理解并推动基因组科学的发展。我们将 AI 系统定义为包括机器学习、深度学习和预测模型。Illumina 致力于根据适用法律和以下指导原则开发和使用人工智能 (AI): 透明度 我们开发的 AI 系统力求可解释和可解读。多样性、非歧视和公平性 我们支持 AI 系统整个生命周期的多样性和包容性,并促进高质量、通用的基因组和医疗保健数据集的创建和可访问性。价值驱动设计 我们设计的人工智能系统进一步实现了我们通过释放基因组的力量来改善人类健康的使命,并反映了我们的价值观,包括高标准的科学卓越性和诚信、负责任的数据管理以及我们的隐私原则、行为准则和人权承诺中所述的价值观。问责制 我们为我们开发的人工智能系统创建了明确的用例,并实施了适当的质量控制和安全措施。我们致力于在我们的人工智能系统中建立人工监督点,并为使用或受我们人工智能系统影响的个人和团体提供反馈机会。

揭示CRISPR/ ... div>的精确维修动态
图1。UMI-DSBSEQ定量单分子测序DSB和修复产品在番茄中的三个靶标。a)时间课的收集:叶肉细胞原生质体是从2-3周大的M82 Solanum Lycopersicum的幼苗中分离出来的。重复的样品在72小时内为72个时间点中的每一个中的每一个制备了200,000个原生质体。CRISPR RNP由PEG介导的转换引入。在提取RNP引入和DNA后,在0、6、12、24、36、48和72小时将样品冷冻。b)UMI-DSBSEQ目标设计:特定于目标序列的引物,与SGRNA目标序列两侧的限制酶位点结合,以创建完整分子(WT或Indel)的可用端,以连接适配器。c)UMI-DSB文库制备:从时间探索收集中提取DNA,其中包含WT(1),未经修复的DSB(2)和包含Indels(3)的完整分子,在体外受到限制,限制了确定目标切割位点的限制酶。通过填充和a添加的最终修复后,由P7 Illumina流量细胞序列和包含i7索引和9BP唯一分子标识符(UMIS)组成的Y形适配器(UMIS)与未经修复的DSB和受限端相连。通过连接介导的PCR进行的靶标特异性扩增,其中一个引物与适配器序列相同,并包含P7 Illumina Tail(橙色)和一个针对靶序列(蓝色)的引物(橙色),带有P5 Illumina Tail(红色)。这会导致SPCAS9切位点和底漆之间的DSB的单端扩增。红色X表示DSB的未捕获端。