XiaoMi-AI文件搜索系统
World File Search Systemcardiomyopathy

扩张型心肌病:病因、机制以及当前和未来的治疗方法
扩张型心肌病的传统定义是,在没有异常负荷条件(如原发性瓣膜疾病)或足以导致心室重塑的严重冠状动脉疾病的情况下,存在左心室或双心室扩张或收缩功能障碍。这一定义被认为过于严格,因为不伴有扩张的左心室运动减少可能是扩张型心肌病的初期表现。扩张型心肌病的病因包括遗传(原发性扩张型心肌病)或后天因素(继发性扩张型心肌病)。后天因素包括感染、毒素、癌症治疗、内分泌病、怀孕、心动过速和免疫介导疾病。5-15% 的获得性扩张型心肌病患者携带可能致病或致病基因变异(即基因突变)。因此,诊断测试和治疗方法应始终考虑遗传和后天因素。该研讨会将重点关注当前的多维诊断和治疗方法,并讨论可能推动未来治疗的潜在病理生理学,旨在修复或替换现有的基因突变,或针对遗传性或获得性扩张型心肌病的特定炎症、代谢或促纤维化驱动因素。

评论文章 - ABC心力衰竭和心肌病
自20世纪初以来,已经描述了导管进行侵入性心脏评估的使用。1,但正是从70年代开始,将肺动脉导管(CAP)用于关键患者边界的血液动力学评估。它在随后的几年中变得很流行,因此在2000年代后期,每年在美国出售了约150万个导管。2研究重症监护病房中关键患者的常规帽效果负面结果的研究或有症状性心力衰竭(CI)的患者具有重力迹象,但没有心脏病性休克,4使他们的使用减少了。然而,在当代时代的心源性休克患者中,使用机械循环辅助装置的患者的最新数据表明,CAP与更大的生存率有关。5

抑制 mTOR 或 MAPK 可改善斑马鱼的 vmhcl/myh7 心肌病
简介心肌病 (CM) 是一组异质性心肌疾病,可分为肥厚性 CM (HCM)、扩张性 CM (DCM) 和限制性 CM (RCM) (1–4)。已鉴定出 CM 的遗传因素,且有 100 多个基因与不同类型的 CM 相关 (5, 6)。已建立动物模型并用于发现关键信号通路和治疗策略。已鉴定出至少 7 条具有治疗潜力的 CM 信号通路,包括丝裂原活化蛋白激酶 (MAPK) 信号转导、mTOR 信号转导、β -肾上腺素能受体信号转导、磷酸二酯酶 5 (PDE5) 信号转导、组蛋白去乙酰化酶 (HDAC) 信号转导、Ca 2+ /钙调蛋白依赖性激酶 II 信号转导和钙调磷酸酶-活化 T 细胞核因子 (Cn-NFAT) 信号通路 (7–9)。例如,mTOR 是一种丝氨酸/苏氨酸蛋白激酶,在调节心肌细胞蛋白质稳态方面起着关键作用 (10–12);通过药理学或遗传学方法部分抑制 mTOR 可对几种类型的心肌病产生心脏保护作用,包括 lamp2 相关 HCM (13)、bag3 相关和层蛋白 A/C 相关 DCM (14, 15) 以及贫血和阿霉素诱发的心肌病 (DIC) (16)。相反,已发现 MAPK 几乎在每种应激和激动剂诱发的肥大刺激下都会激活,并以独特的方式调节心脏离心和向心生长之间的平衡 (17, 18)。 MAPK 的激活会导致离心性肥大并促进肌细胞延长,而抑制细胞外信号调节激酶 (ERK) 通路会减弱对压力超负荷的肥大反应 (19)。MYH7,也称为 β - 肌球蛋白重链,是第一个被确定的 CM 致病基因,后来被确定为约 18% 的 HCM 病例的病因 (20–22)。在人类中,MYH7 与 MYH6 串联位于 14 号染色体上,MYH7 是位于 MYH6 上游的主要成体亚型。在小鼠中,Myh7 和 Myh6 也串联位于 14 号染色体上;然而,上游的 Myh7 基因
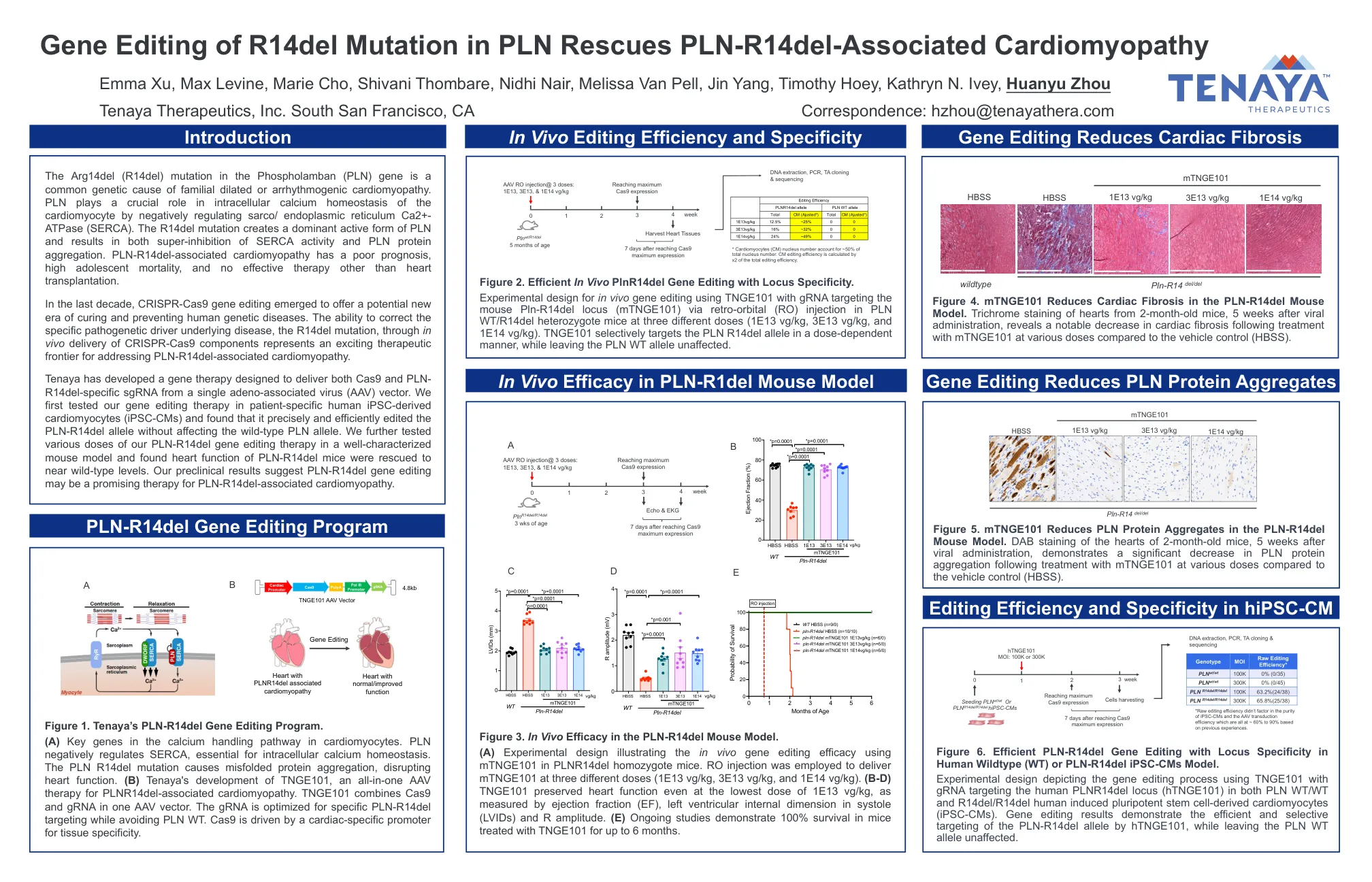
PLN 中 R14del 突变的基因编辑可挽救 PLN-R14del 相关心肌病
Tenaya 开发了一种基因疗法,旨在从单个腺相关病毒 (AAV) 载体传递 Cas9 和 PLN-R14del 特异性 sgRNA。我们首先在患者特异性人类 iPSC 衍生心肌细胞 (iPSC-CM) 中测试了我们的基因编辑疗法,发现它可以精确高效地编辑 PLN-R14del 等位基因,而不会影响野生型 PLN 等位基因。我们进一步在特征明确的小鼠模型中测试了不同剂量的 PLN-R14del 基因编辑疗法,发现 PLN-R14del 小鼠的心脏功能恢复到接近野生型水平。我们的临床前结果表明 PLN-R14del 基因编辑可能是 PLN-R14del 相关心肌病的一种有前途的疗法。

修饰疾病的疗法用于治疗经胸蛋白淀粉样变性心肌病(ATTER-CM)
效果超出了健康和特殊的道德优先事项*,尽管目前可用的治疗方法,但仍有很大的未满足需求。这种情况对于来自卫生保健系统尚未公平服务的种族/族裔的人们具有很大的相关性。这种治疗可能会在照顾者的生活质量和/或追求自己的教育,工作和家庭生活的能力方面取得重大改善。该治疗通过其作用机理或交付方法来改善有效治疗的机会。*在某种程度上,将如何判断任何有效治疗方法的任何有效治疗方法的价值在某种程度上如何判断,并旨在反映特定治疗对患者,护理人员和社会的更广泛影响。有关其他信息,请参阅ICER价值评估框架。ICER鼓励利益相关者在其公众评论中提供有关这些要素的意见。

OPA1处理在小鼠发育中是可赋予的,但在线粒体心肌病中具有保护性
线粒体融合和裂变伴随着压力和代谢需求改变的适应性反应。内膜融合和CRISTAE形态发生取决于视觉萎缩1(OPA1),它以不同的同工型表达,并从膜结合的裂解,长到可溶的短形式。在这里,我们通过生成仅表达一种可裂解的OPA1同工型或不可裂解的变体来分析OPA1同工型和OPA1处理的物理学作用。我们的结果表明,单个可裂解或不可裂解的OPA1同工型的表达可保留胚胎发育和成年小鼠的健康。OPA1处理在代谢和热应力下是可分配的,但可以延长寿命,并预防缺乏OXPHOS缺陷COX10 - / - 小鼠中的线粒体心脏肌病。从机械上讲,OPA1处理的损失会破坏线粒体生物发生和线粒体之间的平衡,从而抑制了Cox10 - / - 心脏中心脏肥大的生长。我们的结果突出了OPA1加工,线粒体动力学和心脏肥大的代谢的关键调节作用。

Znhit1在产后心脏功能和液泡心肌病
引言Ca 2+对于心脏电导传导和收缩至关重要(1,2)。虽然激发反应耦合触发了Ca 2+从肌浆网(SR)释放到通过Ryanodine receptors(Ryrs)到细胞质的,但SR Ca 2+将Ca 2+摄入Ca 2+在很大程度上由SR Ca 2+ -Atpase 2A(Serca 2a(Serca2a)(2,2,2,2,3)。在SR中,Ca 2+由最丰富的Ca 2+结合蛋白(Calsequestrin 2(Casq2)(4)保留。casq1与CASQ2高度同源,这两种蛋白质的作用类似于调节肌肉细胞中Ca 2+的稳态(5)。尽管CASQ1和CASQ2都存在于骨骼肌中,但仅在心肌细胞中发现CASQ2。小鼠遗传学研究表明,尽管SR Ca 2+稳态调节受到破坏,但CASQ1或CASQ2的丧失未能引起致命性心肌病(5)。相反,心肌细胞中具有CASQ2过表达的转基因小鼠患有严重的心肌病,并在16周的时间内过早死亡(6,7)。液泡心肌病是一种罕见但致命的心脏病,具有肌纤维中突出液泡的特征。它通常与溶酶体功能性缺陷有关,包括储存障碍(即富含酸α-葡萄糖苷酶缺乏症)和蛋白质缺乏症(即,Danon疾病,由LAMP2缺乏症引起)(8-10)。然而,经常观察到非散糖体相关的液泡心脏病,其发病机理需要研究(11,12)。染色质复制复合物调节大量基因表达(13)。以前,有报道称SWI/SNF染色质复合物调节心脏发育和产后心脏的生长(14)。例如,SWI/SNF染色质重塑剂的核心成分BRG1促进胚胎心肌细胞增殖并保留心脏分化(15)。在成年小鼠中,心脏应激激活的BRG1诱导病理α -MHC到β -MHC转移,导致肥大(15)。除了SWI/SNF染色质复合物复合物外,哺乳动物还存在其他3种其他染色质重塑剂(ISWI,NURD和INO80/SWR复合物)(13)。但是,与SWI/SNF复合物相比,这3种染色质复合物在产后心脏中的功能仍然未知。含锌手指命中域 - 含有含蛋白的蛋白1(Znhit1;补充表1;本文在线提供的补充材料; https://doi.org/10.1172/jci.insight.1487752ds1),是一个键

评论文章 - ABC 心力衰竭和心肌病
在患有 HF 的患者中,肾上腺素能环路的激活会导致这些受体的过度刺激,重新分配在正常情况下不会对全身循环造成压力的体积,从而产生充血。因此,当建议使用硝酸盐等血管扩张剂时,目的是重新建立最接近静脉床原始容量的容量,并有利于全身充血的流出。如图 2 所示,在血管中心循环压缩模型中,通过降低中心静脉压力,可以优化静脉回流,从而使血液更好地从外周流向心脏。在该方案中,平均系统充盈压控制静脉系统,因此流量优化(Q)的决定因素是应激体积以及右心房压力。因此,恢复电容相当于降低系统屈服点,从而提高效率。 8

南亚印度人中具有临床操作价值的肥厚性心肌病基因
南亚印度人中临床可操作的肥厚性心肌病基因 Vinay J Rao a,b ,理学硕士,Thiagarajan Sairam a ,哲学博士,Andiappan Rathinavel c ,MCh,,Kurukkanparampil Sreedharan Mohanan d ,医学博士,Hisham Ahamed e ,医学博士,Jayaprakash Shenthar f ,医学博士,Perundurai S Dhandapany a,* ,哲学博士。a 心血管发育和疾病机制,干细胞科学和再生医学研究所(DBT-inStem),班加罗尔,印度。b 跨学科健康科学与技术大学,Yelahanka,班加罗尔,印度。c 心血管胸外科系,马杜赖医学院和政府 Rajaji 医院,马杜赖,印度。d 心脏病学系,政府医学院,科泽科德,印度。 e 肥厚性心肌病中心,Amrita 医学科学院,Amrita Viswa Vidyapeetham(Amrita 大学),印度科钦。f 心脏病学系,Sri Jayadeva 心血管科学与研究研究所,印度班加罗尔。* 联系人:Perundurai S Dhandapany;dhan@instem.res.in 摘要背景:原发性肥厚性心肌病 (HCM) 主要是遗传性疾病,在没有其他心脏和全身代谢疾病的情况下导致左心室肥大。目前,关于南亚印度人 (SAI) 中原发性 HCM 临床可操作基因变异的流行率的数据有限,这对于尽量减少对祖先特异性变异的解释差异是必要的。目的:ClinGen 遗传性心血管疾病 (HCVD) 基因管理专家小组根据临床相关性将 HCM 致病基因分为五类:明确、强、中等、有限和有争议。然而,缺乏对 SAI 中这种分类的全面研究。方法:对 335 名原发性 SAI-HCM 患者进行全外显子组测序,包括所有已知的心血管基因和临床可操作的基因类别,以确定它们的等位基因频率。结果:SAI-HCM 外显子组在 335 例中的 119 例 (35.52%) 中揭示了 26 个临床可操作基因中总共 194 个 P/LP 和 VUS。与其他全球 HCM 队列相比,SAI-HCM 队列在 12 个明确类别基因中表现出的变异明显较少(17.33% vs. 41.21%,P = 0.0003)。对于 5 个强/中等基因,SAI-HCM 队列与其他全球 HCM 队列之间无显著差异(2.59% vs. 2.49%,P = 1)。在 21 个有限且有争议的基因中,MYH6 在 SAI-HCM 队列中的变异流行率明显高于其他全球 HCM 队列(5.07% vs. 1.67%,P = 0.0408)。

肥厚型心肌病的遗传学:对临床实践的既定和新兴影响
30%–40% 的肥厚性心肌病病例是由编码心脏肌节蛋白的基因的致病变异引起的。基因检测的主要临床用途是提供诊断确认和促进家庭筛查。它还有助于检测需要不同监测和治疗方法的病因。其他临床应用,包括使用遗传信息来指导风险预测模型,受到建立具有可操作后果的稳健基因型-表型相关性的挑战的限制,但关于罕见和常见遗传变异之间相互作用的新数据,以及针对疾病特异性致病机制的疗法的出现,预示着常规实践中基因检测的新时代的到来。