XiaoMi-AI文件搜索系统
World File Search Systemhypoplasia

MBNL1调节再生和差异化心脏状态之间编程的后出生后切换
MBNL1调节再生和差异性心脏状态之间编程的后产后切换。Logan R.J. Bailey 1,3,4,Darrian Bugg 1,Isabella M. Reichardt 2,C。DessiréeOrtaç1,Jagadambika Gunaje 1,Richard Johnson 5,Michael J. Maccoss 5,Tomoya Sakamoto 6,Tomoya Sakamoto 6,Daniel P. Kelly P. Kelly 6,Michael Regnier 2,7,Michael Regnier 2,7,Jennerifer M.Davis 1,2,7*。 1个实验室医学与病理学,华盛顿大学西雅图,华盛顿大学。 2 Bio Gradineering,华盛顿大学西雅图,华盛顿州。 3分子和细胞生物学,华盛顿大学西雅图,华盛顿大学。 4医学科学家培训计划,华盛顿大学西雅图,华盛顿大学。 5基因组科学,华盛顿大学,西雅图,华盛顿州西雅图6心血管研究所,医学,宾夕法尼亚州宾夕法尼亚大学,宾夕法尼亚州宾夕法尼亚州。 7华盛顿州西雅图市华盛顿大学转化肌肉研究中心 *对应:jendavis@uw.edu摘要发现心肌细胞的成熟度的决定因素和差异化状态的维持对于理解发展和可能重新研究成人哺乳动物心脏作为治疗策略的内源性再生计划至关重要。 在这里,RNA结合蛋白肌肉闪烁1(MBNL1)被确定为心肌细胞分化状态的关键调节剂及其通过对RNA稳定性的转录组控制的再生潜力。 在发育过早过渡的心肌细胞早期,靶向MBNL1过表达,向肥厚性生长,发育不全和功能障碍,而MBNL1功能的丧失会增加心肌细胞周期的进入和通过改变细胞周期抑制剂转录物的扩散。Logan R.J. Bailey 1,3,4,Darrian Bugg 1,Isabella M. Reichardt 2,C。DessiréeOrtaç1,Jagadambika Gunaje 1,Richard Johnson 5,Michael J. Maccoss 5,Tomoya Sakamoto 6,Tomoya Sakamoto 6,Daniel P. Kelly P. Kelly 6,Michael Regnier 2,7,Michael Regnier 2,7,Jennerifer M.Davis 1,2,7*。1个实验室医学与病理学,华盛顿大学西雅图,华盛顿大学。2 Bio Gradineering,华盛顿大学西雅图,华盛顿州。 3分子和细胞生物学,华盛顿大学西雅图,华盛顿大学。 4医学科学家培训计划,华盛顿大学西雅图,华盛顿大学。 5基因组科学,华盛顿大学,西雅图,华盛顿州西雅图6心血管研究所,医学,宾夕法尼亚州宾夕法尼亚大学,宾夕法尼亚州宾夕法尼亚州。 7华盛顿州西雅图市华盛顿大学转化肌肉研究中心 *对应:jendavis@uw.edu摘要发现心肌细胞的成熟度的决定因素和差异化状态的维持对于理解发展和可能重新研究成人哺乳动物心脏作为治疗策略的内源性再生计划至关重要。 在这里,RNA结合蛋白肌肉闪烁1(MBNL1)被确定为心肌细胞分化状态的关键调节剂及其通过对RNA稳定性的转录组控制的再生潜力。 在发育过早过渡的心肌细胞早期,靶向MBNL1过表达,向肥厚性生长,发育不全和功能障碍,而MBNL1功能的丧失会增加心肌细胞周期的进入和通过改变细胞周期抑制剂转录物的扩散。2 Bio Gradineering,华盛顿大学西雅图,华盛顿州。3分子和细胞生物学,华盛顿大学西雅图,华盛顿大学。4医学科学家培训计划,华盛顿大学西雅图,华盛顿大学。5基因组科学,华盛顿大学,西雅图,华盛顿州西雅图6心血管研究所,医学,宾夕法尼亚州宾夕法尼亚大学,宾夕法尼亚州宾夕法尼亚州。 7华盛顿州西雅图市华盛顿大学转化肌肉研究中心 *对应:jendavis@uw.edu摘要发现心肌细胞的成熟度的决定因素和差异化状态的维持对于理解发展和可能重新研究成人哺乳动物心脏作为治疗策略的内源性再生计划至关重要。 在这里,RNA结合蛋白肌肉闪烁1(MBNL1)被确定为心肌细胞分化状态的关键调节剂及其通过对RNA稳定性的转录组控制的再生潜力。 在发育过早过渡的心肌细胞早期,靶向MBNL1过表达,向肥厚性生长,发育不全和功能障碍,而MBNL1功能的丧失会增加心肌细胞周期的进入和通过改变细胞周期抑制剂转录物的扩散。5基因组科学,华盛顿大学,西雅图,华盛顿州西雅图6心血管研究所,医学,宾夕法尼亚州宾夕法尼亚大学,宾夕法尼亚州宾夕法尼亚州。 7华盛顿州西雅图市华盛顿大学转化肌肉研究中心 *对应:jendavis@uw.edu摘要发现心肌细胞的成熟度的决定因素和差异化状态的维持对于理解发展和可能重新研究成人哺乳动物心脏作为治疗策略的内源性再生计划至关重要。在这里,RNA结合蛋白肌肉闪烁1(MBNL1)被确定为心肌细胞分化状态的关键调节剂及其通过对RNA稳定性的转录组控制的再生潜力。在发育过早过渡的心肌细胞早期,靶向MBNL1过表达,向肥厚性生长,发育不全和功能障碍,而MBNL1功能的丧失会增加心肌细胞周期的进入和通过改变细胞周期抑制剂转录物的扩散。此外,与雌激素相关受体信号轴MBNL1依赖性稳定轴对于维持心肌细胞成熟至关重要。根据这些数据,调节MBNL1剂量调整了心脏再生的时间窗口,在该窗口中,增强的MBNL1活性使肌细胞增殖产生了,MBNL1的缺失促进了肌细胞增殖延长的再生状态。总的来说,这些数据表明MBNL1在产后和整个成年期之间充当整个再生和成熟的心肌细胞状态之间的转录组切换。关键字心脏再生,心肌分化,产后发育,转录组稳定,RNA成熟

引用Zhu J-L,Xu X-M,Yin H-Y,Wei J-R和Lyu J(2023),《预测住院时间超过14 da
心室间隔缺陷(VSD)是先天性心脏病的最常见形式,约占先天性心脏病病例的40%(Penny and Vick,2011年)。VSD导致血液分流,从而导致肺部血管的肺部血液循环体积和病理变化增加,这使得患有VSD的儿童特别容易发生肺部感染。随着疾病的发展,当肺循环压力高于全身循环压力时,血液从右侧到左心室的流动会增加左心室的预紧力,从而很容易导致心力衰竭。年轻婴儿的肺泡发育不是完美的,呼吸系统不成熟,并且肺泡II型上皮细胞的合成功能是有效的,导致肺泡表面活性剂的产生较少,因此呼吸功能不成熟。肺动脉症和感染都可能导致氧动脉部分压力降低,这进一步导致呼吸率变化。感染的发生可能是VSD患者长时间住院的危险因素。延长医院可能会进一步增加医院感染的可能性。肺部感染可能会导致肺间隙水肿,导致肺通风和低氧血症减少,最终导致呼吸迅速。某些情况的感染诊断尚不清楚,因此很难确定住院是否延长。因此,我们将本研究的结果设定为住院时间超过14天。但是,呼吸率(RR)是一个易于监控的指标,其测量精度很高,因此它可能具有住院时间的预测价值。一项研究发现,入院率高的呼吸率与疗养院接纳的患者的院内死亡率的增加有关(Myint等,2011)。我们旨在开发一个列图,以评估小儿VSD患者的住院风险超过14天。我们希望临床医生能够根据戒号模型中风险因素的变化进行及时调整治疗方案,以减少小儿VSD患者住院治疗。

药用产品stadex®的名称 - 加上眼滴
其他含有眼药的磷酸盐。由于感染的风险增加,在用皮质类固醇眼睛滴治疗期间,不应佩戴隐形眼镜。全身吸收可以通过压缩液滴安装期间和之后的内侧cantus的泪囊一分钟来减少,这在儿童中尤其建议。氯霉素氯霉素被全身从眼睛中吸收,并且在长期暴露后已被毒性。骨髓性发育不全,包括性贫血和死亡,在局部使用氯霉素后。虽然危险是一种罕见的危害,但在评估使用该化合物的预期收益时,应牢记它。,如果长期或间歇性地使用氯霉素眼滴,则建议在治疗前进行常规的血液剖面,然后以适当的间隔进行检测到任何造血异常。在严重感染中,应通过适当的全身治疗补充氯霉素的局部用途。应避免长时间使用氯霉素眼滴,因为它可能会增加抗性生物的敏化和出现的可能性。如果在治疗过程中出现任何新感染,则应停用抗生素并采取适当的措施。氯霉素应保留仅在特定指示的感染中使用。氯霉素眼滴不能提供针对铜绿假单胞菌和serratia marcescens的足够覆盖范围。在不咨询医生的情况下,不要使用超过5天。如果在2天后没有改善的情况或症状在任何时候恶化,则应寻求医疗建议。如果适用以下任何一项,则应将患者转诊给医生:•视力受到干扰•眼睛内的严重疼痛•恐惧症•眼睛炎症与头皮或脸上的皮疹相关•眼睛看起来多云•瞳孔看起来很不异常•可疑的外国身体在患者中的外国身体应被转介给他们的医生:•以前的医生•con•conjundis•conjuntivis•conjuntivitiv Inter•conjuntivis•conjuntivis•conjuntivis•conjuntivis•conjuntivis•conjuntivis•在过去的6个月中,手术或激光治疗•眼睛损伤•当前使用其他眼滴或眼睛软膏•通过氯霉素眼滴在治疗期间,由于将防腐剂吸收到透镜上,因此不应佩戴软接触透镜,这可能会对晶状体造成损害。建议在眼部感染期间避免所有类型的隐形眼镜。

reln基因
参考•Chang BS,Duzcan F,Kim S,Cinbis M,Aggarwal A,Apse KA,Ozdel O,Atmaca M,Zencir S,Bagci H,Walsh CA.AM J 2007年1月5日; 144b(1):58-63。 doi:10.1002/aj.b.3 •Dazzo E,粉丝,Serioli E,Minvian G,Peltitan P,Striano S,Striana血液A,Radovic S,Spaded A,Uzzau S,Neve A,Giallonard A,消息O,Tosattoc,Tosattoc,Ottman R,Michelusci R,Nobile c Am J Hum Genet。 2015 Jun4; 96:992-1 doi:10.1016。 •fs FS,William,VL,Belmonte PL,Mackinnon DF,FM,Schweer B;国家心理遗传学研究所倡议破坏财团; DePaulo JR,Gershon ES,McMahon FJ,钾肥。 AM J 2010年3月5日; 153b(2):549-5 doi:10.1002/aj.b.31018。 关于PubMed Central•Hong Se,Shugart YY,黄的文章。 DT,Shahwan SA,Grant或Hourihane Jo,Martin ND,Walsh CA。 自湿性隐性lissencephaly伴有人类复发突变的发育不全。 nat Genet。 2000年9月; 26:93-6。 doi:10 1038/79246。AM J2007年1月5日; 144b(1):58-63。 doi:10.1002/aj.b.3 •Dazzo E,粉丝,Serioli E,Minvian G,Peltitan P,Striano S,Striana血液A,Radovic S,Spaded A,Uzzau S,Neve A,Giallonard A,消息O,Tosattoc,Tosattoc,Ottman R,Michelusci R,Nobile c Am J Hum Genet。 2015 Jun4; 96:992-1 doi:10.1016。 •fs FS,William,VL,Belmonte PL,Mackinnon DF,FM,Schweer B;国家心理遗传学研究所倡议破坏财团; DePaulo JR,Gershon ES,McMahon FJ,钾肥。 AM J 2010年3月5日; 153b(2):549-5 doi:10.1002/aj.b.31018。 关于PubMed Central•Hong Se,Shugart YY,黄的文章。 DT,Shahwan SA,Grant或Hourihane Jo,Martin ND,Walsh CA。 自湿性隐性lissencephaly伴有人类复发突变的发育不全。 nat Genet。 2000年9月; 26:93-6。 doi:10 1038/79246。2007年1月5日; 144b(1):58-63。 doi:10.1002/aj.b.3•Dazzo E,粉丝,Serioli E,Minvian G,Peltitan P,Striano S,Striana血液A,Radovic S,Spaded A,Uzzau S,Neve A,Giallonard A,消息O,Tosattoc,Tosattoc,Ottman R,Michelusci R,Nobile cAm J Hum Genet。2015 Jun4; 96:992-1 doi:10.1016。 •fs FS,William,VL,Belmonte PL,Mackinnon DF,FM,Schweer B;国家心理遗传学研究所倡议破坏财团; DePaulo JR,Gershon ES,McMahon FJ,钾肥。 AM J 2010年3月5日; 153b(2):549-5 doi:10.1002/aj.b.31018。 关于PubMed Central•Hong Se,Shugart YY,黄的文章。 DT,Shahwan SA,Grant或Hourihane Jo,Martin ND,Walsh CA。 自湿性隐性lissencephaly伴有人类复发突变的发育不全。 nat Genet。 2000年9月; 26:93-6。 doi:10 1038/79246。2015 Jun4; 96:992-1 doi:10.1016。•fs FS,William,VL,Belmonte PL,Mackinnon DF,FM,Schweer B;国家心理遗传学研究所倡议破坏财团; DePaulo JR,Gershon ES,McMahon FJ,钾肥。AM J 2010年3月5日; 153b(2):549-5 doi:10.1002/aj.b.31018。 关于PubMed Central•Hong Se,Shugart YY,黄的文章。 DT,Shahwan SA,Grant或Hourihane Jo,Martin ND,Walsh CA。 自湿性隐性lissencephaly伴有人类复发突变的发育不全。 nat Genet。 2000年9月; 26:93-6。 doi:10 1038/79246。AM J2010年3月5日; 153b(2):549-5 doi:10.1002/aj.b.31018。关于PubMed Central•Hong Se,Shugart YY,黄的文章。 DT,Shahwan SA,Grant或Hourihane Jo,Martin ND,Walsh CA。自湿性隐性lissencephaly伴有人类复发突变的发育不全。nat Genet。2000年9月; 26:93-6。 doi:101038/79246。勘误:NAT Genet 2001 2月; 27(2):225。引用(https://pubmed.ncbi.nlm.nih.gov/10973257)•Lakatosova S,Ostatnikova D. Reelin及其在脑开发和功能中的复杂参与。Int J Biochem细胞生物。2012年9月; 44(9):1501-4。 doi:10.1016/j.biocel.2012.06.002。Epub 2012 Jun 15。引用于PubMed(https://pubm ed.ncbi.nlm.nih.gov/22705982)•ventruti A,kazdoba tm,niu s,niu s,d' arcangelo G. reeline reeline缺乏症状缺乏症状缺陷,在该分子中的成年构成中的构成综合体的构成。2011年8月25日; 189:32-42。 doi:10.1016/j.neuroscience.2011.05。050.EPUB 2011 JUN 2。引用于PubMed(https://www.ncbi.nlm.nih.gov/pubmed/21 664258)
正常心脏
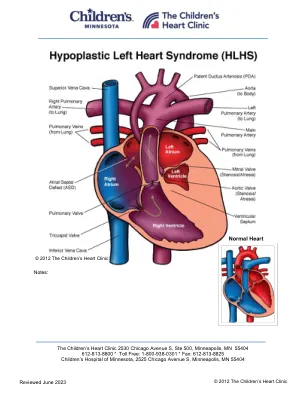
左心脏综合征(HLHS)是指几个密切相关的异常。左心室(LV)是型(小且欠发达)和非功能性的,这意味着右心室必须用作单个抽水室。二尖瓣和/或主动脉瓣的临界狭窄(狭窄)或闭锁(缺勤)以及升主动脉弓和主动脉弓的下降症。在75%的HLHS患者中发生,心室间隔缺陷(VSD)发生在10%中。HLHS发生在所有先天性心脏缺陷的儿童中。身体检查/症状:•心动过速(快速心率),呼吸困难(呼吸困难),肺crack骨,弱外周脉冲和血管收缩在生命的几个小时内很常见。•S2响亮,单身,并且存在绞肉节奏。通常没有心脏杂音。•充血性心力衰竭(CHF)随肝肿大(肝脏增大)而发展。•出生后不久以后不久,氧饱和度通常为90%或较低,并且不会用柔软的氧气改善。诊断:•胸部X射线:中度至重度心脏肿大(心脏增大)和肺静脉会发生。•EKG:演示右心肥大(RVH)。•超声心动图:诊断。有助于确定手术前是否需要心脏导管插入术和血管造影。•心脏导管插入术:有助于评估心脏中的压力。•手术前可能需要进行插管和机械通气。这通常是在心脏导管实验室中完成的。心脏导管通常在第二和第三次姑息手术之前和/或其他干预措施(例如肺动脉(PA)或侧支血管盘绕等其他干预措施)进行。医疗管理/治疗:•对于被诊断为子宫内HLHS的婴儿,建议尽快在三级护理医院转移到心脏重症监护病房,以尽快启动心脏病学评估和医疗干预措施。•前列腺素E(PGE)疗法应在出生后尽快开始,以保留动脉导管的专利,因为这是婴儿唯一的血液流向身体和重要或重要的血液的来源。•新生儿的气囊心房间隔术,没有足够的心房间隔通信,可能有助于改善氧合并在手术前解压缩左心房。•手术修复对于在几个阶段生存是必要的。第一次手术是在生命的第一周内进行的(请参阅Norwood程序)。双向GLENN程序在4-6个月大的年龄之间进行,并在3-4岁之间进行了修改的Fontan程序。•在第一次手术后出院后,婴儿之后是一组心脏促进者,以提供对体重增加和氧气水平的家庭监测。常规心脏病学诊所建议每2-3周一次进行一次,直到婴儿进行第二次心脏手术。

rethymic®(同种异性胸腺组织
可以通过周围血液中幼稚的T细胞的发展观察;在重生治疗后6-12个月之前,不太可能观察到这一点。- 先天性胸骨是一种超稀有的免疫疾病,儿童出生而没有胸腺。婴儿出生的婴儿具有深远的免疫缺陷,并且没有治疗,许多婴儿在2或3岁时死于感染或自身免疫性症状。先天性小没有的确切发病率或患病率尚不清楚,但这种情况会影响男孩和女孩。在美国,每400万婴儿约有17-24名婴儿天生有先天性雌马。 - 先天性胸骨有时被误认为是SCID;两种疾病的患者的T细胞计数非常低。 先天性小没有的小心理和SCID都是原发性免疫缺陷障碍,但它们是2个单独的疾病。 scID是由骨髓造血干细胞功能障碍引起的,而先天性胸肌则与胸腺功能障碍或缺乏有关。 - 先天性胸肌通常发生在患有某些遗传问题的婴儿中,尤其是Digeorge综合征(DGS,有时也称为22Q11.2缺失综合征)。 先天性小心理通常与其他疾病有关,包括coloboma,心脏缺陷,闭锁,choanae,生长和发育迟缓,生殖器性低下,耳朵异常/耳聋(电荷综合征)以及叉子盒蛋白N1(FOXN1)缺乏症。 非环境因素(即) 母亲糖尿病;暴露于酒精,类视黄素或二氯乙酰胺)也与先天性小没有相关。在美国,每400万婴儿约有17-24名婴儿天生有先天性雌马。- 先天性胸骨有时被误认为是SCID;两种疾病的患者的T细胞计数非常低。先天性小没有的小心理和SCID都是原发性免疫缺陷障碍,但它们是2个单独的疾病。scID是由骨髓造血干细胞功能障碍引起的,而先天性胸肌则与胸腺功能障碍或缺乏有关。- 先天性胸肌通常发生在患有某些遗传问题的婴儿中,尤其是Digeorge综合征(DGS,有时也称为22Q11.2缺失综合征)。先天性小心理通常与其他疾病有关,包括coloboma,心脏缺陷,闭锁,choanae,生长和发育迟缓,生殖器性低下,耳朵异常/耳聋(电荷综合征)以及叉子盒蛋白N1(FOXN1)缺乏症。非环境因素(即母亲糖尿病;暴露于酒精,类视黄素或二氯乙酰胺)也与先天性小没有相关。最初,先天性小心理术语被与完全Digeorge Anomaly或Digeorge综合征(CDGS)互换使用,但目前的研究表明,与先天性小没有相关的遗传和非遗传状况不同。- In the 10 prospective, single-center, open-label studies that enrolled 105 patients to evaluate the efficacy of Rethymic, the diagnosis of congenital athymia was based on flow cytometry documenting fewer than 50 naïve T cells/mm3 (CD45RA+, CD62L+) in the peripheral blood or less than 5% of total T cells being naïve in phenotype.- 根据美国过敏,哮喘和免疫学学院(AAAAI; 2017),先天性小没有障碍患者的管理专注于支持护理,以降低感染的风险,直到可以纠正潜在的免疫缺陷为止。与其他原发性免疫缺陷类似,一旦怀疑先天大自然,建议将医院的新生儿与空气过滤系统(HEPA,LAF)相反。- 除隔离外,先天性胸肌患者还应开始抗菌预防,以预防细菌,病毒和真菌感染。- B细胞功能通常在这些患者中降低,因此应接受免疫球蛋白替代。人类免疫球蛋白用于静脉内或皮下给药的制剂是影响体液免疫系统的原发性免疫缺陷疾病患者的治疗基石。鉴于人类免疫球蛋白的潜在风险和固有的稀缺性,因此有必要仔细考虑其指示和管理。- 造血干细胞移植(HSCT)已在先天性小心肌患者中进行,其成功相对较少。HSCT后的生存率较低,据报道,在大约50%的患者中,无胸腺患者中有重大不良事件。HSCT后无胸腺患者的免疫重建也很差,没有明确的幼稚T细胞再生的证据。

MIRAGE 综合征的基础研究,旨在开发治疗策略 MIRAGE 综合征是最近发现的一种遗传性疾病,
对 MIRAGE 综合征进行基础研究以开发治疗策略 MIRAGE 综合征是一种最近发现的遗传性疾病,其特点是六个主要特征,包括骨髓发育不良、感染、生长受限、肾上腺发育不全、生殖器表型和肠病。“MIRAGE”是这六个特征的首字母缩写。MIRAGE 综合征是由 SAMD9 突变引起的,该突变编码一种功能未知的蛋白质。MIRAGE 综合征是一种罕见/难治性疾病。日本仅发现 11 名患者。MIRAGE 综合征是一种危及生命的疾病,事实上,超过一半的患者在 2 岁前死亡。我们开展“对 MIRAGE 综合征进行基础研究以开发治疗策略”的研究旨在获得有关 MIRAGE 综合征的基本知识和见解,从而有助于开发治疗方法。成海聪(国立儿童保健与发育研究所分子内分泌科主任)建立了 MIRAGE 综合征的 HEK293 细胞模型,研究人员可以通过该模型重现患者细胞的生长受限情况。利用该模型,他测试了大约 1,500 种之前鉴定的小化合物,以寻找治疗 MIRAGE 综合征的潜在药物。然而,在初步筛选中尚未发现任何有效的化合物。目前,SAMD9 的功能在很大程度上尚不清楚。鉴定 SAMD9 的功能对于阐明 MIRAGE 综合征的分子机制至关重要。为此,成海聪和金仓耕介(东京医科大学分子病理学系助理教授)开始了两种基于细胞的实验。一种是蛋白质组学筛选。在该实验中,以上述 MIRAGE 综合征的 HEK293 细胞模型的细胞提取物为对象,用抗体偶联树脂捕获 SAMD9,并寻找与 SAMD9 结合的分子。已确定了几种候选分子,目前正在验证中。另一个是基因组学筛选。Narumi 和 Kanekura 使用基因编辑技术应用了一种新的基因敲除筛选方法,现在正试图确定负责 SAMD9 功能的生物学途径。基于细胞的方法对于研究 MIRAGE 综合征的分子和细胞水平发病机制是有效的。另一方面,这些方法不适合阐明器官和身体水平的发病机制。它需要对 MIRAGE 综合征患者进行深入表征,并重现该疾病的动物模型。为了对患者进行深入分析,Tomonobu Hasegawa(庆应义塾大学医学院儿科教授)与日本儿科内分泌学会和日本新生儿健康与发展学会一起开始了全国性的 MIRAGE 综合征调查。这项调查将有助于找到更多患者,并将有助于阐明该综合征的临床表现。此外,为了建立MIRAGE综合征的动物模型,木下昌人(京都大学农学研究科应用生物科学系助理教授)和谷口义人(预防医学和公共卫生系教授)正在培育基因工程的青鳉(Medaka)。石井智宏(庆应义塾大学医学院儿科助理教授)也在培育基因工程小鼠。今年,靶向载体的构建已经完成。这些实验将在明年建立突变动物系。

未校正的证明
doi:10.4274/jcrpe.galenos.2024.2024-7-11病例报告,由于InsGeneenández等人的新变体,由于新变体而导致的胰岛素需求高的永久性新生儿糖尿病。在Ins Gene Johana Andrea BoteroHernández1,GinaGonzález-Valencia 1,Vanessa Suarez 2,Vanessa Suarez 2,Gabriel Del Castillo 2,Gabriel del Castillo 2 1 1 1 1 1 1 1哥伦比亚大学哥伦比亚2.关于这个话题? 导致永久性新生儿糖尿病的三种最常见的致病变异涉及ABCC8,KCNJ11和INS基因。 后者负责10-20%的病例,并导致可变的临床行为,与宫内生长限制,Mody型糖尿病以及永久性或短暂的新生儿糖尿病有关。 这项研究添加了什么? 本研究报告了INS基因中的一种新型的致病变异,未在数据库中记录。 与ABCC8和KCNJ11变体不同,INS变体对磺酰脲治疗没有反应,需要胰岛素进行血糖控制,在母乳喂养患者中构成挑战。 它突出了对早期分子诊断支持的临床方法的需求。 抽象的新生儿糖尿病是一种不经常出现的疾病,可能以短暂性,永久性或综合症的形式出现。 最常见的是涉及ABCC8,KCNJ11和INS基因的致病变异引起的。 由于先前未报告的INS基因变体,描述了一个永久性糖尿病的新生儿,概述了诊断复杂性,治疗性干预措施以及相关的临床挑战。 GinaGonzález-Valencia,Antioquia大学。在Ins Gene Johana Andrea BoteroHernández1,GinaGonzález-Valencia 1,Vanessa Suarez 2,Vanessa Suarez 2,Gabriel Del Castillo 2,Gabriel del Castillo 2 1 1 1 1 1 1 1哥伦比亚大学哥伦比亚2.关于这个话题?导致永久性新生儿糖尿病的三种最常见的致病变异涉及ABCC8,KCNJ11和INS基因。后者负责10-20%的病例,并导致可变的临床行为,与宫内生长限制,Mody型糖尿病以及永久性或短暂的新生儿糖尿病有关。这项研究添加了什么?本研究报告了INS基因中的一种新型的致病变异,未在数据库中记录。与ABCC8和KCNJ11变体不同,INS变体对磺酰脲治疗没有反应,需要胰岛素进行血糖控制,在母乳喂养患者中构成挑战。它突出了对早期分子诊断支持的临床方法的需求。抽象的新生儿糖尿病是一种不经常出现的疾病,可能以短暂性,永久性或综合症的形式出现。最常见的是涉及ABCC8,KCNJ11和INS基因的致病变异引起的。由于先前未报告的INS基因变体,描述了一个永久性糖尿病的新生儿,概述了诊断复杂性,治疗性干预措施以及相关的临床挑战。GinaGonzález-Valencia,Antioquia大学。GinaGonzález-Valencia,Antioquia大学。新生儿具有对称的宫内生长限制,他们出现了与酮症或感染性无关的严重高血糖。他有很高的胰岛素要求,并且没有对磺酰脲管理做出反应。抗胰岛素和抗ISLET胰腺抗体为阴性。 遗传测序显示INS基因中的纯合错义变体(c.3g> a,p.met1ile),该变体以前尚未在文献中报道过。 及时对新生儿糖尿病的分子诊断可以优化管理策略,从而减轻对生长,神经发育和降血糖发作的长期影响。 关键字:新生儿糖尿病;新生;胰岛素基因。 麦德林。 哥伦比亚gina.ginzalezv@udea.edu.co 05.08.2024 11.11.2024 EPUB:20.12.2024简介新生儿糖尿病(NDM)是一种罕见的遗传状况,普遍存在,患病率是1/300,000至1/300,000的活性,vary berthers berthers,vary vary nive vary nistical,vary nistical soprication(vary)(vary)。 在欧洲,估计患病率在1/90,000至1/300,000的活产之间(3)。 在高血缘关系的地区,例如安纳托利亚(土耳其东南地区)和中东,患病率可能会增加到1/21,000至1/48,000个活产(1)。 NDM的发作通常发生在生命的前六个月内,尽管已记录了9至12个月的较晚案例。 自身免疫性糖尿病也应通过针对谷氨酸脱羧酶(GAD),胰岛素,锌转运蛋白和酪氨酸磷酸酶进行负抗体测试来排除。 没有血缘家族历史。抗胰岛素和抗ISLET胰腺抗体为阴性。遗传测序显示INS基因中的纯合错义变体(c.3g> a,p.met1ile),该变体以前尚未在文献中报道过。及时对新生儿糖尿病的分子诊断可以优化管理策略,从而减轻对生长,神经发育和降血糖发作的长期影响。关键字:新生儿糖尿病;新生;胰岛素基因。麦德林。哥伦比亚gina.ginzalezv@udea.edu.co 05.08.2024 11.11.2024 EPUB:20.12.2024简介新生儿糖尿病(NDM)是一种罕见的遗传状况,普遍存在,患病率是1/300,000至1/300,000的活性,vary berthers berthers,vary vary nive vary nistical,vary nistical soprication(vary)(vary)。在欧洲,估计患病率在1/90,000至1/300,000的活产之间(3)。在高血缘关系的地区,例如安纳托利亚(土耳其东南地区)和中东,患病率可能会增加到1/21,000至1/48,000个活产(1)。NDM的发作通常发生在生命的前六个月内,尽管已记录了9至12个月的较晚案例。自身免疫性糖尿病也应通过针对谷氨酸脱羧酶(GAD),胰岛素,锌转运蛋白和酪氨酸磷酸酶进行负抗体测试来排除。没有血缘家族历史。当血浆葡萄糖水平超过150-250 mg/dL时,应怀疑NDM的诊断,尤其是在排除其他高血糖的其他潜在原因之后,包括败血症,低出生体重或与早产相关的并发症,以及诸如苯乙糖蛋白,苯甲酸,糖皮质激素,Iropropic或High dextropic inspropic或High dextropics(4-4)的药物(等过早相关的并发症)。NDM的关键生化特征是降低基底胰岛素水平和C肽的水平(4,5)。迄今为止,具有不同的遗传模式的NDM发病机理已与40多个基因有关。These genes affect insulin synthesis, action, and secretion by altering beta cell development (aplasia and pancreatic hypoplasia), increasing beta cell destruction by apoptosis or protein misfolding with consequent endoplasmic reticulum stress due to retained proteins, and altering beta cell membrane depolarization leading to failure in the extrusion of synthesized insulin into the circulation (3)。最常见的基因是ABCC8,KCNJ11和INS。后者位于11p15.5染色体上,造成6.7至18%的病例,并导致与宫内内生长限制和Mody-type糖尿病相关的可变临床行为(1,7)。本报告提出了因INS基因中新型致病性变异的新生儿糖尿病病例,其胰岛素要求非常高。我们旨在强调分子诊断在建立及时有效管理中的作用。病例报告了两天的男性新生儿被送入新生儿单位。患者的母亲18岁,正在接受她的第一次怀孕。遵循足够的婴儿是在妊娠35周通过剖宫产部分分娩的,其出生体重为1,310克,长度为44厘米,头圆周长为29厘米(长度为Z分数-1.05,体重 - 成年Z分数-2.71的体重-2.71,head-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age-age z得分。新生儿最初表现出适当的适应性,但在生命的前两天内发展出临床替代性,其特征是严重的贫血需要输血,间歇性鼻窦心动过缓,超声心动膜造影正常,持久性超血糖症和血糖水平持久性超血糖水平达到574 mg/dl。排除了传染病学,家族病史和药物诱导的高血糖症后,考虑了新生儿糖尿病的诊断。最初以0.07 U/kg/h的速率进行静脉胰岛素输注的患者进行管理。随后,每12小时以0.8 u/kg/的最大剂量与柔性方案中施用的胰岛素Aspart一起,以每12小时的最大剂量为0.8 u/kg/。由于NDM中ABCC8和KCNJ11突变的高流行,进行了磺酰尿素的治疗试验,但这无法改善血糖控制。