XiaoMi-AI文件搜索系统
World File Search Systempromotes

腺苷信号传导抑制红细胞生成并促进髓样分化
造血是由骨髓中造血干细胞(HSC)产生所有血细胞的过程。促红细胞生成和颗粒状是造血的两个主要分支,分别是红细胞(RBC)和中性粒细胞的生产。虽然红细胞和髓样分化均来自相同的常见髓样祖细胞(CMP),但这两个过程之间的相互作用是复杂的,并且由不同的内在和外在因素紧密地策划,这些因子调节了祖细胞对一个细胞谱系或另一个细胞谱系或另一个细胞谱系的组合。1个末端红细胞生成和粒状植物发生在红细胞岛上,这些岛屿是骨髓中的专门微环体,该微晶体由中央宏观噬菌体组成,周围环绕着红细胞和中性粒细胞前体。2这些结构构成了独特的细胞微环境,并且通过提供必需的营养素,去除细胞碎片以及分泌细胞因子和生长方面来支持细胞增殖和分化至关重要。3越来越多的证据表明,在这些壁ches中发生平衡的微环境提示以及代谢物的运输和信号,还有其他

IIA类HDAC抑制剂TMP269促进BMP ... -orca
中脑乳突多巴胺能神经元的变性是帕金森氏病(PD)的病理标志。化合物的外围递送以阻止或减慢这种多巴胺能变性是一个关键的治疗目标。组蛋白脱乙酰基酶(HDAC)酶(关键表观遗传调节剂)在PD模型中表现出治疗前景。但是,由于有几类HDAC(Classi-IV),因此特定类别的抑制对于确保目标特异性很重要。在这里,我们检查了IIA类HDAC抑制剂TMP269的神经保护潜力。我们表明,TMP269在SH-SY5Y细胞和培养的大鼠腹脑中脑多巴胺能神经元中受到6-羟基多巴胺(6-OHDA)诱导的神经突损伤的影响。我们发现TMP269上调了SH-SY5Y细胞中神经营养因子BMP2和BMP-SMAD依赖性转录信号传导,这对于其针对6-OHDA诱导损伤的神经保护作用是必不可少的。此外,周围连续输注0.5 mg/kg的TMP269通过迷你渗透泵7天,减少了纹状体6-OHDA给药引起的前肢损伤。TMP269还保护了Nigra及其纹状体6-OHDA诱导的神经变性的底层中的多巴胺能神经元,并防止了6-OHDA在Vivo中的IBA1阳性微胶质细胞的数量增加,IBA1阳性微胶质细胞的数量增加。TMP269还防止了BMP2,PSMAD1/5和乙酰化组蛋白3水平的6-OHDA诱导的降低,并且它反转了6-OHDA诱导的核HDAC5在本次Nigra的多巴胺能神经元中核HDAC5的增加。这些数据增加了越来越多的证据体系,即IIA类特异性HDAC抑制剂可能是感兴趣的外围递送的药理学剂,其目的是在PD中进行神经保护。

蝾螈大脑中的神经元激活促进脊髓再生
(未经同行评审认证)是作者/资助者。保留所有权利。未经许可不得重复使用。此预印本的版权所有者此版本于 2024 年 12 月 20 日发布。;https://doi.org/10.1101/2024.12.19.629459 doi:bioRxiv preprint

Top2a 通过 PRC2 和 H3K27me3 促进社会行为的发展
人们对社会性的胚胎发育了解甚少。我们筛选了 1120 种已知药物,发现胚胎抑制拓扑异构酶 II α (Top2a) 会导致斑马鱼出现持久的社会缺陷。在小鼠中,产前 Top2 抑制会导致社交互动和交流缺陷,而这些行为与自闭症的核心症状有关。斑马鱼 Top2a 突变导致一组基因下调,这些基因高度富含与人类自闭症相关的基因。Top2a 调节的和自闭症相关的基因组都具有多梳抑制复合物 2 (PRC2) 的结合位点,PRC2 是一种负责 H3K27 三甲基化 (H3K27me3) 的调节复合物。此外,这两个基因组都高度富含 H3K27me3。抑制 PRC2 成分 Ezh2 可挽救 Top2 抑制引起的社会缺陷。因此,Top2a 是进化保守途径的关键组成部分,该途径通过 PRC2 和 H3K27me3 促进社会行为的发展。

治疗诱导的转分化促进胶质瘤生长,不依赖于 EGFR 信号传导
7 病理学和实验室医学系,北岸大学医院和长岛犹太医学中心,诺斯韦尔健康中心,莱克萨克塞克斯,霍夫斯特拉/诺斯韦尔唐纳德和芭芭拉扎克医学院,纽约州 11042,美国
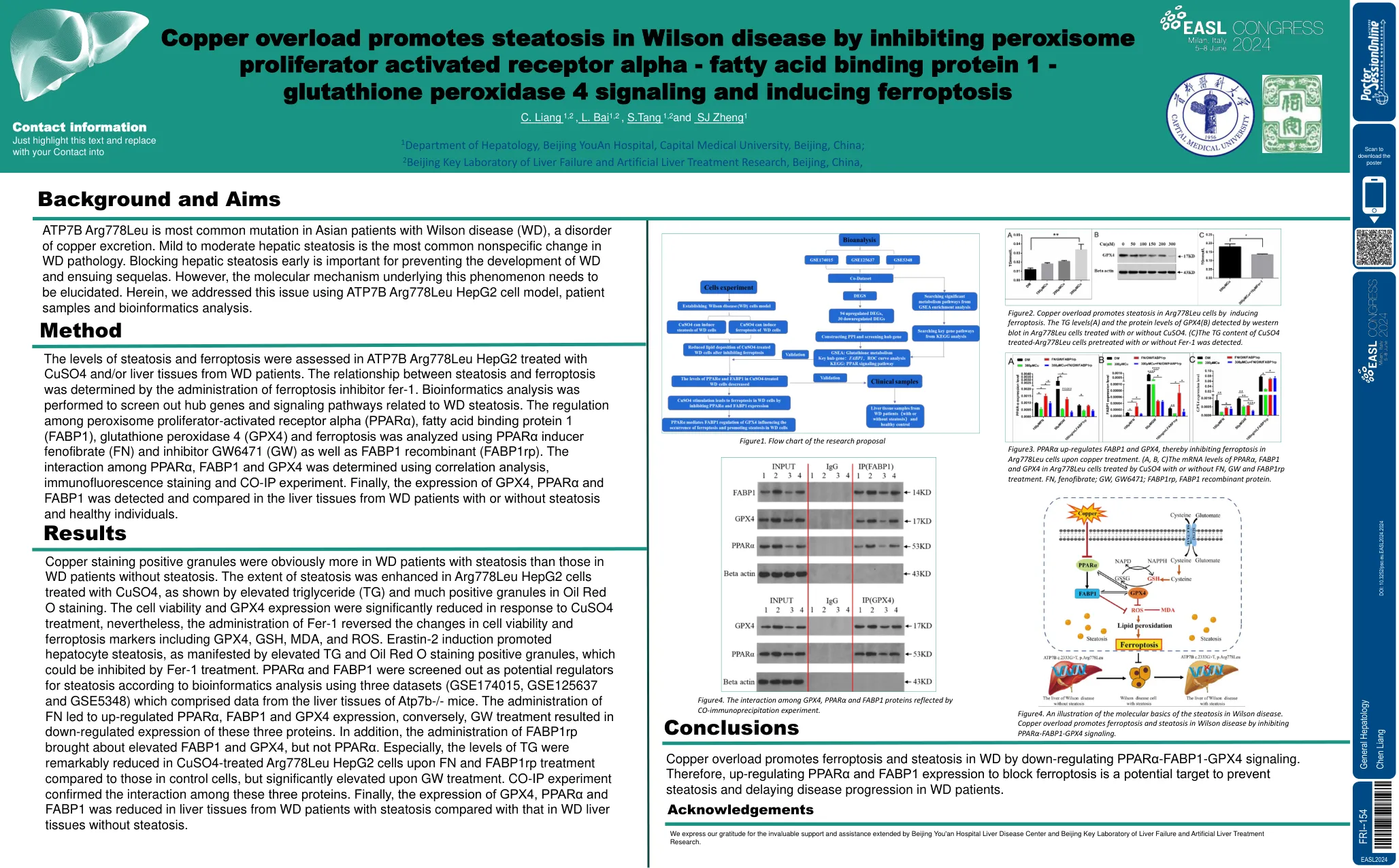
铜超载通过抑制过氧化物酶体促进威尔逊疾病中的脂肪变性
铜染色阳性颗粒显然比没有脂肪变性的WD患者的脂肪变性患者更多。在用CUSO4处理的ARG778LEU HEPG2细胞中,脂肪变性的程度得到了增强,如甘油三酸酯升高(TG)和油红色O染色中的阳性颗粒所示。对CUSO4处理的响应,细胞活力和GPX4表达显着降低,但是,FER-1的给药逆转了细胞生存能力和包括GPX4,GSH,GSH,MDA和ROS在内的细胞生存力和铁凋亡标记的变化。erastin-2诱导促进了肝细胞脂肪变性,如TG和油红O染色阳性颗粒所表现出的,这可以受到FER-1处理的抑制。PPARα和FABP1作为脂肪变性的潜在调节剂进行筛选,该数据集包含来自ATP7B-小鼠的肝脏组织的数据。FN的施用导致了上调的PPARα,FabP1和GPX4表达,相反,GW处理导致这三种蛋白的表达下调。此外,FABP1RP的给药带来了FABP1和GPX4的升高,但没有PPARα。,与对照细胞中的FN和FABP1RP处理后,经过FN和FABP1RP处理后CUSO4处理的ARG778LEU HEPG2细胞的TG水平明显降低,但在GW处理后却显着升高。co-IP实验证实了这三种蛋白质之间的相互作用。最后,与没有脂肪变性的WD肝组织相比,WD患者的肝组织中GPX4,PPARα和FABP1的表达降低。

KEAP1 通过抑制 NSCLC 中的 PD-L1 表达来促进抗肿瘤免疫
记录版本:该预印本的一个版本于 2024 年 2 月 27 日在《细胞死亡与疾病》上发表。已发表的版本请参阅 https://doi.org/10.1038/s41419-024-06563-3 。

剪切应力调节促进猪心内膜的促炎反应
。cc-by-nc-nd 4.0国际许可证(未经同行评审证明)获得的是作者/资助者,他授予Biorxiv授予Biorxiv的许可,以永久显示预印本。它是制作

循环外DNA促进高风险髓母细胞瘤
OWE S. Xinlian Zhang 26,Horrad Y. Wechsler-Reya 3.8,Vineet Baphna 4.29,Jill P. 2.29.30&Luke Chavez 2.3.14.2

辅助负荷促进以目标为导向的拉伸反射增益调节
自愿运动在执行前需要做好准备。人们已在整个中枢神经系统中观察到了准备活动,最近在人类周围神经系统(即肌梭)的一级神经元中也发现了准备活动。感觉器官中出现的变化表明,拉伸反射增益的独立调节可能是运动准备的重要组成部分。本研究的目的是进一步研究人类受试者优势上肢的短延迟拉伸反射反应 (SLR) 和长延迟拉伸反射反应 (LLR) 的准备调节。具体来说,我们研究了不同的目标参数(目标距离和方向)如何影响目标导向伸手的背景下拉伸反射增益的准备调节,以及任何此类调节是否取决于准备持续时间和背景负荷的方向。我们发现目标距离只会产生很小的反射增益变化。相比之下,SLR 和 LLR 增益都根据目标方向受到强烈调节,从而促进即将到来的自愿运动。当准备延迟足够长(> 250 毫秒)且同向肌肉未负重时,这种以目标为导向的 SLR 和 LLR 增益调节会出现或增强 [即,当背景负荷首次施加在同向肌肉动作方向(辅助负荷)时]。结果进一步支持了伸手准备中相对缓慢进化的过程,该过程可能通过独立控制肌腱运动神经元来调节反射性肌肉僵硬。这种控制可以增强自愿的目标导向运动,并在同向肌肉未负重时被触发或增强。