XiaoMi-AI文件搜索系统
World File Search SystemrRNA

长度16S rRNA测序
KINNEX 16S rRNA试剂盒将扩增的16S扩增子作为输入,并输出一个可进行测序的库,与标准的FL 16S库相比,该库将导致多达12倍的吞吐量增加。Kinnex 16S套件基于多路复用阵列测序(MAS-SEQ)方法(Al'khafaji等,2024),用于FL 16S扩增子(图3A)。结果明显更高,并且对高精度,成本效率的FL 16S测序的测序需求显着降低,每个PACBIO SMRT细胞的多重能力高达1,536个扩增子样品。我们在各种样本中测试了Kinnex 16S rRNA套件,包括模拟社区标准,凳子,唾液,植物,土壤,废水,废水和拭子(皮肤,口腔,阴道和兽医伤口)。然后,我们使用用户友好的生物信息学管道HIFI-16-Workflow分析了数据,该管道为FL 16S HIFI读取提供了快速Q-to-Report分析解决方案(图3B)。我们还检查了读取深度对样本类型中检测到的物种数量的影响(图4)。

禾本科植物中的选择性 rRNA 基因抑制
核仁显性 (ND) 是 35-48S rDNA 基因座的选择性表观遗传沉默。在异源多倍体中,它通常表现在细胞遗传学水平上,即从一个或多个进化祖先遗传下来的核仁组织区 (NOR) 失活。禾本科植物在生态和经济上是最重要的陆生植物群之一,它们经常通过杂交和多倍化事件进化。在这里,我们从细胞遗传学、分子和基因组学的角度回顾了这个单子叶植物科中 ND 现象的共同特征和独特特征。我们重点介绍了使用异源四倍体模型禾本科植物 Brachypodium hybridum 取得的最新进展,其中 ND 通常发生在种群水平,并且我们介绍了解读 NOR 核心阵列结构特征的现代基因组方法。

Neoadjuvant Nivolumab加化学疗法,然后在HPV阴性头颈癌中反应分离化的化学放疗治疗。
背景:肠道菌群是一个复杂的生态系统,在人类健康和疾病中起着至关重要的作用。但是,肠道菌群与烧伤引起的肠道损伤之间的关系尚不清楚。肠粘膜层对于维持肠道稳态和提供针对细菌侵袭的生理障碍至关重要。本研究旨在研究肠道菌群对烧伤后肠道粘液的合成和降解的影响,并探索烧伤损伤的潜在治疗靶标。方法:采用了修改的组织病理学分级系统来研究烧伤损伤对小鼠结肠组织和肠道粘液屏障的影响。随后,使用16S核糖体RNA测序分析燃烧后第1至10天的肠道菌群的变化。基于此,对在第1、5和10天收集的样品进行了宏基因组测序,以研究与粘液相关的微生物群的变化并探索潜在的潜在机制。结果:我们的发现表明粘液屏障被破坏,小鼠烧伤后第3天发生细菌易位。此外,从烧伤后的第1到第3天,小鼠中的肠道菌群显着破坏,但随着疾病的进展,逐渐恢复到正常。特别是,在燃烧后的第1天,与粘蛋白降解相关的共生和致病细菌的丰度显着增加,但第5天的丰度恢复了正常。相反,与粘蛋白合成相关的益生菌丰度在相反的方向上发生了变化。进一步的分析表明,在烧伤损伤后,能够降解粘液的细菌可能利用糖苷水解酶,叶叶菌和内膜分解粘液层,而合成粘液的细菌可能通过促进短链脂肪酸的产生来帮助恢复粘液层。结论:烧伤损伤导致肠结肠粘液屏障和肠道菌群的营养障碍。一些共生和致病性细菌可能通过糖苷水解酶,叶酸,内膜等参与粘蛋白降解。益生菌可以提供短链脂肪酸

16S rRNA基因测序的见解
背景:口腔健康取决于复杂的口腔微生物环境。此可变的口服微生物组包括牙齿,牙龈和舌头上的微生物种群。遗传学,食物,口腔卫生和健康状况都影响了这些微生物生态系统。这种生态学是敏感的,中断可能导致口腔失调。口腔粘膜炎和种植体周围感染经常是由于这种失衡而引起的。 这种广泛的分析检查了口腔病原体,例如链球菌,乳酸杆菌和卟啉念珠菌。 16S rRNA基因测序用于研究这些细菌如何引起口腔疾病。 这种强大的分子方法照明了口服微生物动力学。 这项研究通过检查这些致病性微生物与口腔健康之间的复杂相互作用来照亮疾病的发展。 本文还强调需要使用尖端的科学方法来治疗口腔疾病。 它描述了这些工具如何改变了口腔病理研究。 这项研究的发现对于改善治疗和预防方法至关重要。 评论强调口腔卫生并鼓励其他研究。 牙科治疗可能会变得更加个性化和有针对性,从而改善了口腔健康管理。口腔粘膜炎和种植体周围感染经常是由于这种失衡而引起的。这种广泛的分析检查了口腔病原体,例如链球菌,乳酸杆菌和卟啉念珠菌。16S rRNA基因测序用于研究这些细菌如何引起口腔疾病。这种强大的分子方法照明了口服微生物动力学。这项研究通过检查这些致病性微生物与口腔健康之间的复杂相互作用来照亮疾病的发展。本文还强调需要使用尖端的科学方法来治疗口腔疾病。它描述了这些工具如何改变了口腔病理研究。这项研究的发现对于改善治疗和预防方法至关重要。评论强调口腔卫生并鼓励其他研究。牙科治疗可能会变得更加个性化和有针对性,从而改善了口腔健康管理。

长度16S- ITS-23S rRNA操纵子序列
收到2024年2月2日; 2024年5月7日接受;于2024年6月7日发布:1 Doherty应用微生物基因组学,微生物学和免疫学系,墨尔本大学Peter Doherty感染与免疫学研究所,792 Elizabeth Street,Melbourne VIC 3000,澳大利亚澳大利亚墨尔本街792号; 2爱尔兰科克摩尔帕克的Teagasc食品研究中心; 3爱尔兰科克大学科克大学科克大学科克大学的APC微生物组和微生物学院; 4 Vistamilk SFI研究中心,爱尔兰科克Teagasc Moorepark。*信件:John G. Kenny,John。Kenny@teagasc。IE关键字:Amplicons;数据库;长阅读测序;微生物组;纳米孔; rRNA。缩写:COV,变异系数; ESV,精确的序列变体; Grond,基因组衍生的核糖体操纵子数据库; GTDB,基因组分类数据库; IQR,四分位数范围;它的内部转录垫片; NR,非冗余; ONT,牛津纳米孔技术; RRN,16S-ITS-23S rRNA操纵子; rRNA,核糖体RNA; SD,标准偏差; Taxlca,集群中所有序列的最低祖先; Taxmaj,最低的分类学等级,其中所有序列中的所有序列都具有简单的多数协议; Taxrep,集群代表序列的源基因组分类学; UMIS,唯一的分子标识符。数据语句:文章或通过补充数据文件中提供了所有支持数据,代码和协议。本文的在线版本可以使用两个补充表。001255©2024作者

真核ITS和rRNA扩增子的全长HIFI测序
与简短的读数相比,覆盖范围仅限于ITS1或ITS2区域,HIFI读取的读数涵盖了全真菌其区域的全长。这包含800 bp,如果包括18s和/或28S基因以跨越操纵子,则可以提供约5 kb的扩增子。以及更保守的rRNA基因,这些区域允许在物种水平上降低序列相似性和更高分辨率的分类信息(Tedersoo等,2018; Tedersoo等人,2021年,图2)。除了产生更高的分类价值外,HIFI全长扩增子测序的成本与短阅读的部分扩增子测序相当。在本申请注释中,我们提供了从复杂社区DNA样品中扩增全长真核ITS和rRNA区域的一般指导。

NextSeqtm 1000和NextSeq 2000
文库,并比较了NextSeq 2000和Miseq系统之间的测序性能。在图书馆准备过程中,使用IDT进行Illumina DNA/RNA UD索引设置A到D,使用户可以生成384 16S库。在NextSeq 1000/2000 P2 300M试剂套件(600个周期)上运行384 16S库,具有标准SBS化学或NextSeq 1000/2000 P2 Xleap-SBS™试剂盒(600个循环),每样品可用于分类级别的每样品,以产生100,000至200,000的读取。用户可以在NextSeq 1000/2000 P2 300M试剂盒(600个循环)和400m读取的NextSeq 1000/2000 P2 XLEAP-SBS Reagent套件(600 Cycles)上生成300m总读取。NextSeq 1000/2000 P1 XLEAP-SBS试剂盒(600个循环)和NextSeq 1000/2000 P2 Xleap-SBS试剂盒(600个周期)的测序运行时间为34小时。相比,Miseq Reagent Kit V3(600个周期)的Miseq系统上的测序运行时间〜56小时。
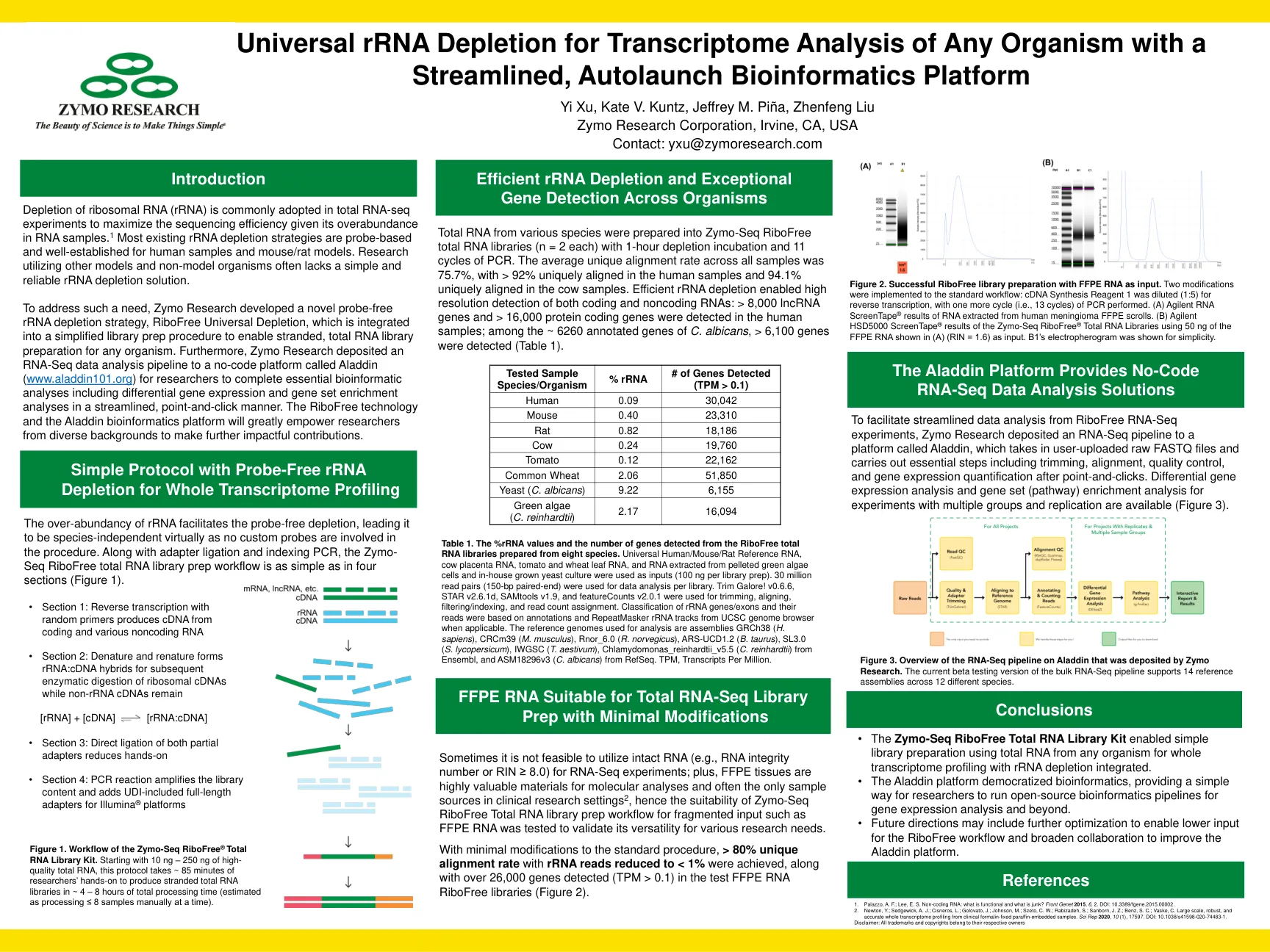
通用rRNA耗竭用于任何有机体的转录组分析
表1。从八个物种制备的无核能总RNA文库中检测到的%rRNA值和基因数量。通用的人/小鼠/大鼠参考RNA,牛胎盘RNA,番茄和小麦叶RNA以及从沉淀的绿藻细胞中提取的RNA和内部成年酵母菌培养物用作输入(每位图书馆100 ng)。每个库的数据分析使用了3000万读对(150 bp配对)。修剪大奖!v0.6.6,Star v2.6.1d,Samtools v1.9和farmaturecounts v2.0.1用于修剪,对齐,过滤/索引和读取计数分配。RRNA基因/外显子的分类及其读取是基于UCSC基因组浏览器的注释和retoMasker rRNA轨道的基础。 用于分析的参考基因组是组件GRCH38(H。SAPIENS),CRCM39(M。MUSCULUS),RNOR_6.0(R。NORVEGICUS),ARS-UCD1.2(B. Taurus),SL3.0,SL3.0,SL3.0(S. lycopersicum),IWGSC(iwgsc),IWGSC(iwgsc),iwgsc(iwgscim),chlamans,C。c. c. c. c. c. c. c. anasen nasunson。 Reinhardtii)来自Ensembl和Refseq的ASM18296V3(C. albicans)。 tpm,百万分的成绩单。RRNA基因/外显子的分类及其读取是基于UCSC基因组浏览器的注释和retoMasker rRNA轨道的基础。用于分析的参考基因组是组件GRCH38(H。SAPIENS),CRCM39(M。MUSCULUS),RNOR_6.0(R。NORVEGICUS),ARS-UCD1.2(B. Taurus),SL3.0,SL3.0,SL3.0(S. lycopersicum),IWGSC(iwgsc),IWGSC(iwgsc),iwgsc(iwgscim),chlamans,C。c. c. c. c. c. c. c. anasen nasunson。 Reinhardtii)来自Ensembl和Refseq的ASM18296V3(C. albicans)。tpm,百万分的成绩单。

16S rRNA ngs粪便细菌菌群的测序...
目标。这项研究的目的是使用16S rRNA基因的下一代测序(NGS)来表征并可能区分健康和肥胖马的下肠道(粪便)细菌。方法。这项研究涉及7匹马(4匹马和3匹母马),年龄8-17岁:乌克兰鞍品种1-4匹马(马1运动马匹rebus,10 Y.O.,马匹2马匹2种马santes,15 Y.O.,15 Y.O.,15 Y.O.),重量吃水的5匹马(种马Tsyhan,8 Y.O.)和非透明马6和7(Mare Sne-Zhynka,10 Y.O.,Mare Rumba 12 Y.O.)马匹2、4、5和7是肥胖,马1、3和6是健康的。所有马匹都保留在州生物技术大学的马术中心,乌克兰教育与科学部(乌克兰哈尔基夫)。根据制造商的说明,使用Purelink微生物组DNAPuriÞ阳离子试剂盒(Invitrogen,USA)提取直肠粪便样品的总DNA。准备了细菌16S rRNA的库,我们使用了16S rRNA条形码试剂盒1-24(美国牛津纳米波尔)。为了净化所获得的库,磁性颗粒核元素清理和尺寸选择(Macherey-Nagel,德国根据推荐的快速测序放大器的建议协议 - 16S条形码(SQK-16SS024)(测序套件的手册)。这些条件基于Fujiyoshi等人(2020)中所述的16S rRNA基因扩增阳离子的标准方案,并确保细菌DNA跨各种群分类群的稳健扩增。结果。结论。细菌门的代表(Syn.肌动杆菌),纤维杆菌,小叶虫 - 螺旋杆菌(Syn.螺旋体),杆菌,富公司(Syn.芽孢杆菌),planctomycetota,verrucomicrobiota(Syn.verrucomicrobia),念珠菌Melainabacteria,kiritimatiellota和proteeobacteria(Syn.假单胞菌)。占主导地位的门是坚硬的,其份额是所有检测到的门的50%至82%。与杆菌的数量相比,健康马匹和肥胖马之间的数量差异很大。在健康马1,3和6中,这是企业和肥胖的马2,4,5和7的2.5、3.4和2.9倍,它是8.6、8.2、7.6和5.7倍。与杆菌相比,坚硬的人数在健康马匹和肥胖马之间发生了显着变化。在健康的马1、3和6中,牢固的数量分别为2.5、3.4和2.9倍,而在肥胖的马2、4、5和7中,牢固的数量分别为8.6、8.2、7.6、7.6和5.7倍。在肥胖的马匹2、4、5和7中观察到蛋白杆菌的数量增加,范围为25%至37%,而在健康运动马1、3和6中,蛋白质的水平在1.07至3.43%之间,这对于健康动物的微生物组典型。在研究的马匹粪便中检测到低水平的放线菌(分杆菌):健康运动马3分别为0.09%,健康运动马3分别为0.09%,健康马匹6分别为0.15%。相比之下,肥胖的马2、4、5和7的水平分别从0.21%到0.48%。重要的是要注意,放线菌的门还包括BiÞ多杆菌属,在所研究的任何动物中均未检测到。在乌克兰第一次,我们对七个不同年龄,性别和品种的七匹马的下肠道(粪便材料)的细菌菌群进行了测序。在肥胖马的粪便中,细菌的细菌占主导地位(天细菌粉,粉状,裂缝),尤其是来自振荡性螺丝素和lachnospileceae的家族,并伴随着细菌的降低细菌(fcylumberimteroidota)(FC-fcbe)(FC-fc-

比较16S rRNA基因测序研究的subgingival Microbiota 1
样品比比利时(p = 1.3 x 10 -5,p = 2.0 x 10 -4),智利(p = 4.0 x 10 -4,p = 1.5 x 10 -2)和166