XiaoMi-AI文件搜索系统
World File Search Systemclones

应用人工智能来加速和降低风险......
与所有科学和工业领域一样,人工智能 (AI) 有望在未来几年对抗体的发现产生重大影响。抗体的发现传统上是通过一系列实验步骤进行的:动物免疫、相关克隆的筛选、体外测试、亲和力成熟、动物模型体内测试,然后是不同的人性化和成熟步骤,产生将在临床试验中进行测试的候选物。该方案存在不同的缺陷,使整个过程非常危险,流失率超过 95%。计算机方法的兴起,包括人工智能,已逐渐被证明能够以更强大的过程可靠地指导不同的实验步骤。它们现在能够覆盖整个发现过程。在这个新领域的参与者中,MAbSilico 公司提出了一种计算机流程,可以在几天内设计抗体序列,这些序列已经人性化并针对亲和力和可开发性进行了优化,大大降低了风险并加快了发现过程。

BIOS 11174生物学研究经验
在许多情况下,在许多情况下,物种的生存率将受到土地利用,气候和大气化学改变的前所未有的组合的影响。预期这些趋势很困难,但是洞察力可能来自人口对过去的环境扰动的反应。麦克拉克兰实验室(McLachlan Lab)收集了人口变化的记录,并使用数学模型将其与环境和生物过程联系起来,这些模型将居住在大西洋盐沼泽(Schoenoplectus americanus)的基础植物物种(Schoenoplectus Americanus)连接到改变环境变量(盐度,洪水,洪水和大气CO 2)。令人难以置信的是,美国链球菌的种子可以追溯到150年,仍然可行,这些独特植物的克隆在温室实验中已经传播。遗传实验已经确定并证实了这些“复活”植物是目前在盐沼中发现的植物的不同基因型。因此,该实验室对可以追溯到一个世纪以来的环境变化的进化反应有连续的记录。

一个多功能的托管病毒衍生的纳米颗粒,其靶向癌细胞并内化为
随着合成生物学的努力变得更加雄心勃勃,活细胞中预定义功能的工程需要越来越准确的工具。此外,遗传构建体的表型性能的表征需要细致的测量和广泛的数据获取,以实现进食数学模型和沿设计构建测试生命周期的匹配预测。在这里,我们开发了一种遗传工具,可以简化高通量转座子插入测序(TNSEQ):携带HIMAR1 Mariner Transposase System的PBLAM1-X质粒载体。这些质粒源自Mini-TN5转座子矢量PBAMD1-2,并按照标准欧洲矢量体系结构(SEVA)格式的模块化标准构建。为了展示它们的功能,我们分析了60个土壤假单胞菌putida kt2440克隆的测序结果。新的PBLAM1-X工具已经包含在最新的SEVA数据库版本中,在这里我们使用实验室自动化工作流程描述其性能。

基因编辑针对DUX4聚腺苷酸化信号
摘要:Facioscapulohumeral营养不良(FSHD,OMIM:158900,158901)是成年人中最常见的Dys-Tropherphy,到目前为止,还没有治疗。已经表征了疾病的不同基因座,它们都导致Dux4蛋白的异常表达,这会损害肌肉的功能,最终导致细胞死亡。在这里,我们使用基因编辑来试图通过靶向其poly(a)序列永久关闭Dux4表达。我们在FSHD成肌细胞上使用了类似转录激活剂样效应子核酸酶(TALEN)和CRISPR-CAS9核酸酶。测序了150多个Topo克隆,仅观察到4%的indels。重要的是,在其中2个中,Dux4 poly(a)信号在基因组水平上被消除,但由于使用了非典型上游poly(a)信号序列,仍会产生DUX4 mRNA。这些实验表明,在基因组水平上靶向DUX4 PA可能不是FSHD治疗的适当基因编辑策略。

TN5标记并将寡核酸转移到单链DNA中,以进行链特异性RNA测序
哺乳动物细胞中的遗传筛选通常专注于功能丧失方法。为了评估额外基因拷贝的表型后果,我们使用了辐射杂种(RH)细胞的大量分离分析(BSA)。,我们构建了六个RH细胞池,每个池由约2500个独立克隆组成,并将池放置在带有或没有紫杉醇的培养基中。低通序测序鉴定859个生长基因座,38个紫杉醇基因座,62个相互作用基因座和3个基因座,用于跨基因组的明显限度,用于线粒体丰度。分辨率被测量为约30 kb,接近单基因。差异性特性,反驳了平衡假设。此外,在RH池中,人类丝粒的保留增强表明,这些染色体元素的功能解剖方法是一种新的方法。对RH细胞的合并分析显示出高功率和分辨率,应该是哺乳动物遗传工具包的有用补充。

用于高通量筛选的标准化转座子工具
随着合成生物学研究的规模越来越大,在活细胞中设计预定义功能需要越来越精确的工具。此外,遗传构建体表型性能的表征需要细致的测量和广泛的数据采集,以便在设计-构建-测试生命周期中为数学模型提供信息并匹配预测。在这里,我们开发了一种简化高通量转座子插入测序 (TnSeq) 的遗传工具:携带 Himar1 Mariner 转座酶系统的 pBLAM1-x 质粒载体。这些质粒源自 mini-Tn5 转座子载体 pBAMD1- 2,并按照标准欧洲载体结构 (SEVA) 格式的模块化标准构建。为了展示它们的功能,我们分析了 60 个土壤细菌 Pseudomonas putida KT2440 克隆的测序结果。新的 pBLAM1-x 工具已经包含在最新的 SEVA 数据库版本中,我们在这里使用实验室自动化工作流程描述了它的性能。

癌细胞状态:人类肿瘤单细胞RNA序列十年的经验教训
人类肿瘤是复杂的生态系统,由多种遗传克隆和在复杂肿瘤微环境中进化的恶性细胞状态组成。单细胞RNA-sequencing(SCRNA-Seq)提供了一种剖析这种复杂生物学的策略,并使我们在过去十年中了解肿瘤生物学的能力进行了革命。在这里,我们反映了人类肿瘤中SCRNA-SEQ的这一十年,并高光照这些研究收集的一些强大的见解。我们首先关注鲁棒定义癌细胞态及其多样性的计算方法,并突出了跨癌症类型的基因表达内肿瘤内异质性(EITH)的一些最常见的模式。然后,我们在定义和命名此类EITH程序的领域中讨论了歧义。最后,我们重点介绍了关键的发展,这些发展将促进未来的研究并在临床环境中更广泛的这些技术实施。
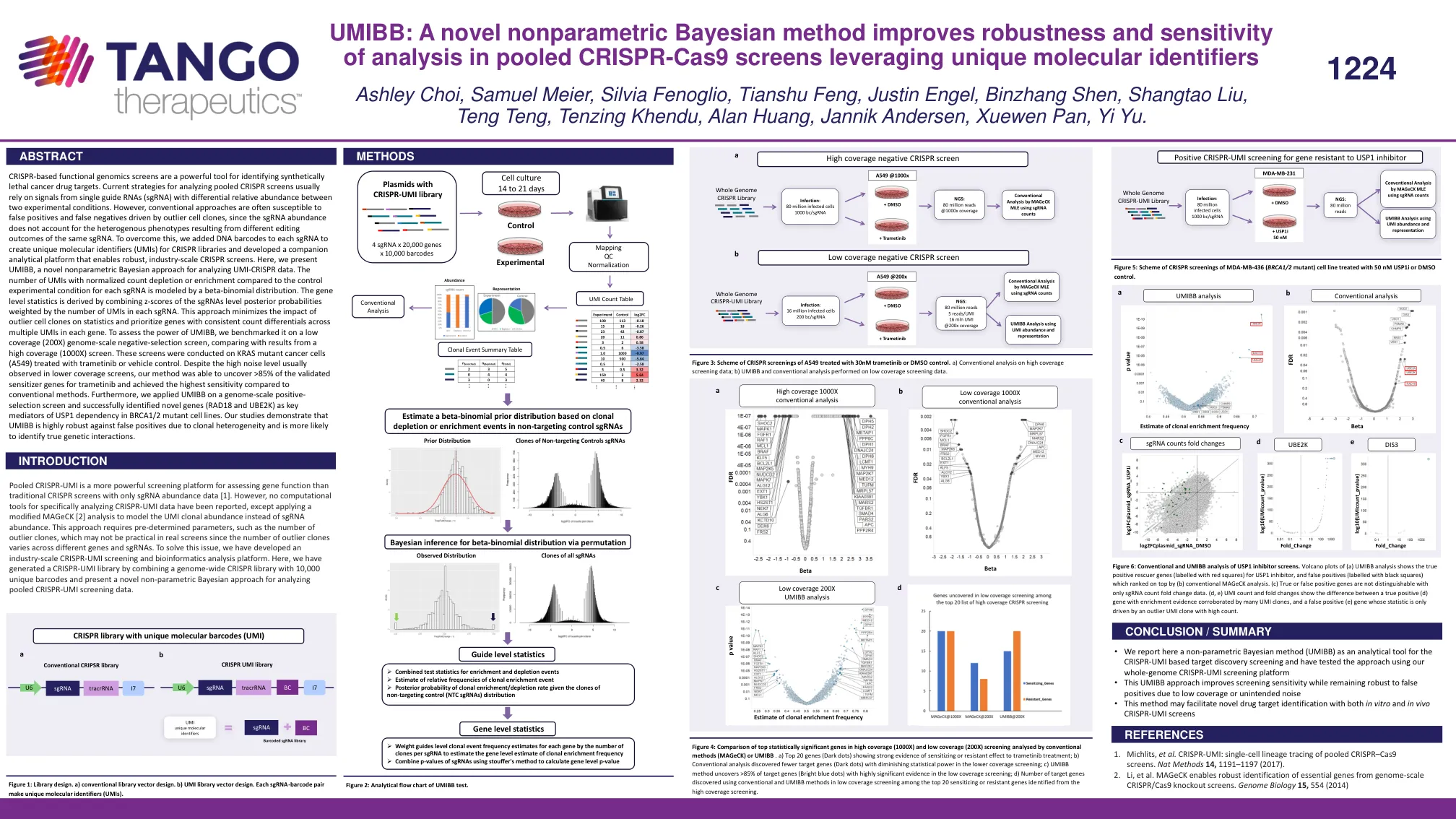
UMIBB:一种新的非参数贝叶斯方法提高了稳健性和灵敏度
基于 CRISPR 的功能基因组学筛选是识别合成致死癌症药物靶点的有力工具。目前分析汇集的 CRISPR 筛选的策略通常依赖于来自在两种实验条件下具有不同相对丰度的单个向导 RNA (sgRNA) 的信号。然而,传统方法通常容易受到由异常细胞克隆驱动的假阳性和假阴性的影响,因为 sgRNA 丰度不能解释由相同 sgRNA 的不同编辑结果导致的异质表型。为了克服这个问题,我们在每个 sgRNA 中添加了 DNA 条形码,以创建 CRISPR 文库的唯一分子标识符 (UMI),并开发了一个配套的分析平台,以实现强大的工业规模 CRISPR 筛选。在这里,我们介绍了 UMIBB,一种用于分析 UMI-CRISPR 数据的新型非参数贝叶斯方法。与每个 sgRNA 的对照实验条件相比,具有标准化计数消耗或富集的 UMI 数量由 beta-二项分布建模。基因水平统计数据是通过将 sgRNA 水平后验概率的 z 分数与每个 sgRNA 中 UMI 的数量加权而得出的。这种方法最大限度地减少了异常细胞克隆对统计数据的影响,并优先考虑每个基因中多个 UMI 之间计数差异一致的基因。为了评估 UMIBB 的功效,我们在低覆盖率(200X)基因组规模负选择筛选上对其进行了基准测试,并与高覆盖率(1000X)筛选的结果进行了比较。这些筛选是在用曲美替尼或载体对照处理的 KRAS 突变癌细胞(A549)上进行的。尽管在较低覆盖率筛选中通常会观察到高噪音水平,但我们的方法能够发现 >85% 的曲美替尼已验证的致敏基因,并且与传统方法相比实现了最高的灵敏度。此外,我们将 UMIBB 应用于基因组规模的正向选择筛选,并成功确定了新基因(RAD18 和 UBE2K)是 BRCA1/2 突变细胞系中 USP1 依赖性的关键介质。我们的研究表明,UMIBB 对克隆异质性导致的假阳性具有很高的稳健性,并且更有可能识别真正的遗传相互作用。

化脓性链球菌 Cas9 核糖核蛋白递送,用于在锥虫和利什曼原虫中进行高效、快速和无标记的基因编辑
图 1 布氏锥虫 PCF 中的 GFP 失活。(a)对组成性表达胞浆 eGFP 的布氏锥虫进行荧光流式细胞术分析。在用 20 μ g(无 Cas9、Cas9/gRNA GFP1、Cas9/gRNA GFP2、Cas9/gRNA GFP3)或 60 μ g(Cas9/gRNA GFP2)来自 IDT 的 RNP 复合物转染后 24 至 72 小时随时间监测 GFP 荧光,条形图显示用不同向导转染后 72 小时 GFP 阴性细胞的百分比(n = 3)。采用 Prism 软件进行统计分析,采用 t 检验(非配对、正态分布、参数检验和双尾)。显着性水平(p 值)用星号表示。 (b)上图显示了允许 e Sp Cas9 在大肠杆菌中表达的质粒的示意图。蓝色框表示蛋白质 N 端和 C 端的两个多组氨酸序列,红色框表示 TEV 和肠激酶 (EK) 蛋白酶的切割位点,灰色框表示三个核定位信号 (NLS),黑色框表示 FLAG 表位的三个重复,橙色框表示 e Sp Cas9 编码序列。下图显示了在用来自 IDT 或实验室纯化 (Lab) 的 RNPs 复合物 (无 Cas9、20 μ g Cas9/gRNA GFP2、40 μ g Cas9/gRNA GFP2、40、60 和 80 μ g Cas9/gRNA GFP2) 转染后 72 小时监测的表达 GFP 的 T. brucei 的荧光流式细胞术分析。(c)不再表达 GFP 的克隆中 GFP 基因的一部分的序列比较。该序列仅显示 GFP2 向导 RNA 所针对的区域。灰色框(H1 和 H2)突出显示可能用于 MMEJ 修复的同源区域。由实验室纯化的 Cas9 失活产生的序列和来自商业 Cas9 的序列分别标记为 Lab 和 IDT。下面显示了 Dc6 和 Ba10 克隆的相应色谱图(置信区间 95%— p 值样式:0.1234 (ns);0.0332 (*);0.0021 (**);0.0002 (***);< 0.0001 (****))。

快速隔离广泛中和mab
SARS-COV-2大流行强调了对研究体液免疫反应并鉴定具有诊断性和治疗潜力的单克隆抗体(MAB)的迫切需求。记忆B细胞(MBC),对自适应免疫的关键,在抗原重新接合时会产生高亲和力抗体。虽然单细胞高通量测序已彻底改变了抗体曲目研究,但它具有关键的局限性:无法同时确定抗原结合特异性和免疫球蛋白基因序列,并且高资源需求限制了低回能设置中可访问性的限制。在这里,我们提出了一个具有成本效益的单细胞培养(SCC)平台,可以对人类MBC曲目进行全面分析,包括表位特异性响应,交叉反应性研究和MAB分离。使用SARS-COV-2康复和疫苗接种样品,我们优化了使用NB21馈线细胞,R848和IL-2刺激的MBC SCC,与散装培养物相比,具有高克隆效率和30倍富集的抗原特异性MBC。在592个孤立的mAb中,有52.7%的人对武汉菌株尖峰蛋白表现出特异性,靶向受体结合结构域(RBD)的27.9%,15.4%的N末端结构域(NTD)和56.7%的其他区域,可能是S2域。比较分析显示出不同的交叉反应性模式:所有抗尖峰mAB中有40.5%识别所有经过测试过的SARS-COV-2变体(Wuhan,Beta,Delta,Gamma和Omicron Ba.2),而29.6%的人仅显示四个变体,不包括Omicron Ba.2,仅显示了四个变体的认可;还鉴定出56个单应力反应性mAb(14.9%)。所需的克隆以进行重组单键产生。值得注意的是,所有筛选和中和测定都直接用培养上清液进行,从而消除了对大规模测序和转染的需求。SCC平台还启用了无偏的免疫球蛋白曲目分析,揭示了收敛的V区域重排,包括公共V3-30和V3-53/V3/V3-66抗体与先前的SARS-COV-2研究一致。在伪病毒中和测定中验证了两个具有广泛中和潜力的公共靶向克隆。这个简化的平台在7天之内提供了同时提供抗原特异性的MAB隔离,V区域测序和功能研究,从而赋予低资源环境中的研究人员能够解决全球健康不平等,并增强对未来大流程学的准备。