XiaoMi-AI文件搜索系统
World File Search System缓冲液
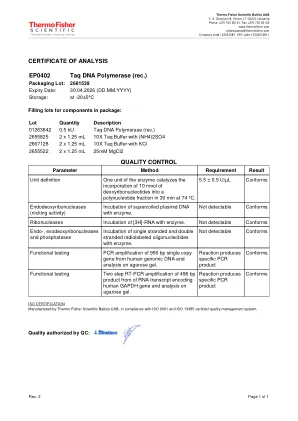
EP0402 TAQ DNA聚合酶(rec。) Invitrogen转染产品 Lentipool V2人CRISPR库 - 用户指南 实现特定于目标效率-truecut-hifi-cas9-
批量描述01263642 0.5 KU TAQ DNA聚合酶(rec。)2655825 2 x 1.25 ml 10x TAQ缓冲液(NH4)2SO4 2667128 2 x 1.25 ml 10x TAQ缓冲液与KCl 2655522 2 x 1.25 ml 25mm 25mm 25mm mgcl2

克隆实验手册
1) 将大肠杆菌培养液(高拷贝质粒:2-10 ml)离心(12,000 x g,30秒),弃上清,得到沉淀。 ↓ ②加入150 μl A1 buffer(加RNase A),涡旋悬浮细胞。 ↓ ③加入250μl A2缓冲液,颠倒混合5次左右,静置2分钟。 [裂解] ↓ ④ 加入350 μl A3缓冲液,颠倒混匀,直至液体由蓝色变为完全无色。检查是否没有蓝色残留,然后离心(12,000 x g,3 分钟)。 ↓ ⑤将上清液转移到NucleoSpin® Plasmid EasyPure 柱中,离心(1,000-2,000 × g,30 秒)。 [结合] ↓ ⑥ 加入450 μl AQ缓冲液(+EtOH)并离心(12,000 × g,1分钟)。 [洗涤/干燥] ↓ ⑦向柱中加入50 μl AE缓冲液,室温下放置1分钟。 ↓ ⑧ 离心(12,000×g,1分钟)回收质粒溶液。 [洗脱]

组蛋白去乙酰化酶抑制剂(伏立诺他)
• 方法 2(亲和纯化) 1. 洗涤 4 o C(冰上)用洗涤缓冲液洗涤珠子 3 次。 2. 添加样品溶液 将 1000ul HeLa 细胞提取物添加到珠子中。 3. 反应 4 o C 孵育 240 分钟。 4. 洗涤 除去样品溶液。4 o C(冰上)用洗涤缓冲液洗涤珠子 3 次。 5. 洗脱 伏立诺他洗脱: - 添加 40ul 洗脱缓冲液并重新悬浮珠子。在 98 o C 下煮沸 5 分钟并除去珠子。 伏立诺他洗脱: + 在洗涤缓冲液中加入 30ul 25mM 伏立诺他并重新悬浮珠子。将管放在冰上一小时,通过间歇轻拍使结合的蛋白质洗脱。旋转后,磁性分离。将上清液转移到新的干净管中98 o C 煮沸 5 分钟。 6. 通过 SDS-PAGE 和银染分析样品

Genematrix通用DNA/RNA/蛋白质纯化试剂盒
注意6·通过添加能够破坏二硫键键的还原剂(例如ß-甲醇(ß -me)或二硫代硫醇(DTT))的减少剂,从而污染了污染的RNass。为了促进二硫键的还原,使用前,每1 mL缓冲液DRP加入10 µLß -ME。添加ß -ME后,DRP缓冲液保持稳定1个月。使用前,每1 ml缓冲液在使用前,在RNase无rNase无水中添加10 µl [1 m] DTT的毒性但更昂贵的替代品。dtt在缓冲区DRP中不稳定,因此不得存储DTT-供应的DRP缓冲液等分试样。[1 M] DTT储备溶液在RNase无水酶中的工作等分试样必须存储在-20°C下,以保持稳定性。设置[1 M] DTT储备溶液(MW = 154.25 g mol -1),溶解1.54 g DTT每10 ml RNase无rNase无水,并将其存储在等分试样中以进行一次使用。

我的泰铢必须是ute。 •它应包含96井。 •50件...
14。基因组DNA纯化试剂盒•试剂盒应适用于哺乳动物培养基细胞,细菌,酵母和组织样品进行基因组DNA净化。•套件必须适合纯化GDNA,终点PCR,QPCR和NGS的库准备应用。•套件与旋转结肠技术一起使用。•套件含量应为50个反应。•组织裂解缓冲液(12mL),GDNA结合缓冲液(24ml),GDNA洗涤缓冲液(1 8ml),GDNA 80ml(14ml),自旋柱(50件),自旋收集管(50片),proteinase K(0.55ml)和RNase A(0.55ml)和RNase A A和RNase A (0.17毫升)应具有满足感。15。热启动TAQ DNA聚合酶•酶200单位(5000单位/ml)和0.04ml的0.04ml。•基于适体的抑制作用。•最多5 kb的目标应适用于LAN的放大。•酶热启动功能。•5'皮瓣必须具有IndonuctLease活性。•除了酶外,还应应为1 .5ml 10x标准TAQ反应缓冲液。
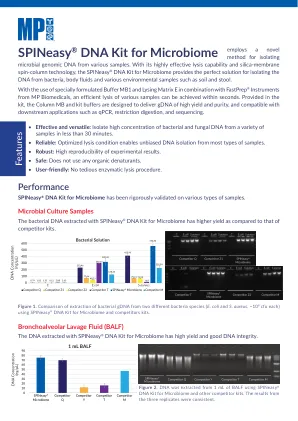
mpure细菌DNA提取试剂盒Spineasy®DNA套件用于微生物组
使用特殊配方的缓冲液MB1和裂解矩阵E与MP BioMedicals的FastPrep®仪器结合使用,可以在几秒钟内实现各种样品的有效裂解。在套件中提供的列MB和套件缓冲液旨在提供高产量和纯度的GDNA,并与QPCR,限制消化和测序等下游应用兼容。

用磷光铂(II)配合物的DNA寡核苷酸的位点特异性金属化用轻型缓冲液控制DNA纳米版
合成的DNA/RNA链是出色的工程材料,用于开发纳米版和纳米机器,可以在传感中找到应用,1个药物输送,2个成像3和分子运输。4 Watson-Crick – Frank-Lin碱基配对的高可编程性,以及相互作用的可逆性以及将其用作多功能分子支架的可能性,使合成DNA特别适合设计精确的纳米级结构。2 B,5,6基于DNA的纳米器件通常是通过理性设计的 - 可识别特定分子输入(例如核酸,7个小分子8或蛋白质)的特定分子输入的核酸域而开发的。9通过多种外源刺激(包括温度10

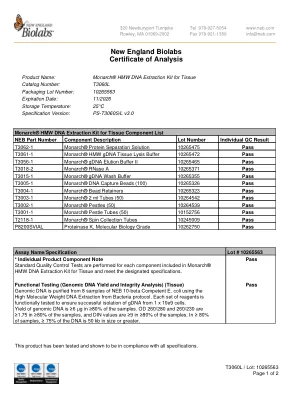
新英格兰生物实验室分析证书
NEB 部件编号 组件描述 批号 单个 QC 结果 T3062-1 Monarch® 蛋白质分离溶液 10265475 通过 T3061-1 Monarch® HMW gDNA 组织裂解缓冲液 10265472 通过 T3056-1 Monarch® gDNA 洗脱缓冲液 II 10265465 通过 T3018-2 Monarch® RNase A 10265371 通过 T3015-1 Monarch® gDNA 洗涤缓冲液 10265355 通过 T3005-1 Monarch® DNA 捕获珠 (100) 10265326 通过 T3004-1 Monarch® 珠子固定器 10265323 通过 T3003-1 Monarch® 2 ml 试管 (50) 10264542 通过 T3002-1 Monarch® 研杵(50) 10264539 合格 T3001-1 Monarch® 研杵管 (50) 10152756 合格 T2118-1 Monarch® 旋转收集管 10245009 合格 P8200SVIAL 蛋白酶 K,分子生物学级 10262750 合格

分子生物学实验室使用 GelRed 染料对琼脂糖凝胶中的核酸进行染色
在凝胶制备过程中,使用浓度为 1.5% 的 TBE 缓冲液 (Tris-Borate-EDTA) 琼脂糖作为核酸电泳的基质。采用了两种不同的方法,以适应染色技术。为了使用 GelRed® 进行电泳后染色,在不添加任何类型的染料的情况下制备凝胶,然后将染料与浓度为 1:9 的上样缓冲液混合。使用该混合物将样品上样到琼脂糖凝胶中,使用 2ul 缓冲液 + GelRed® 和 6ul 扩增的 PCR 产物。然而,为了染色预电泳凝胶,通过预染色将溴化乙锭掺入琼脂糖中。这是通过在融化后将 0.5 μg/mL 的 EtBR 添加到 100 mL 琼脂糖中来实现的。在这两种方法中,电泳技术都是在以下条件下进行的