机构名称:
¥ 1.0
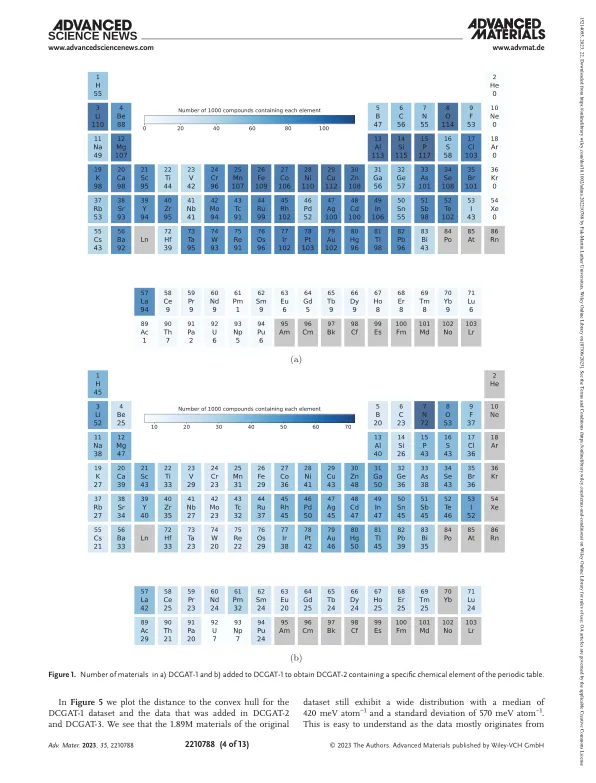
系统探索了跨越数百万材料的化学空间,寻找具有针对特定技术应用的量身定制特性的化合物。[1-4]当前,预测化学计量的最有效方法是扫描固定晶体结构原型的组合空间。[5-7]在这种方法中,用于估计材料是否可以实验合成的关键材料特性是总能量,或者更多的是与热力学稳定性凸壳的能量距离。[6,8–17]典型地,给定化学成分和晶体结构原型(即,勇敢的晶状体的组合和一组占用的Wyckoff位置)进行几何优化,例如,使用密度功能理论的某些风味(DFT),并将其与之相比。[18,19]凸壳上的化合物(或接近它)进行表征,如果它们具有有趣的物理或化学特性,则提出了用于实验合成的。尽管如此,合成反应是极其复杂的过程,而与凸船的距离与合成性相关,但不足以决定是否可以在实验上访问材料。最近的几部作品通过直接预测最佳合成条件或合成概率来解决此问题。[20–25]
高级材料-2023 -Schmidt -Machine -Learning- ...
主要关键词




相关文件推荐