机构名称:
¥ 1.0
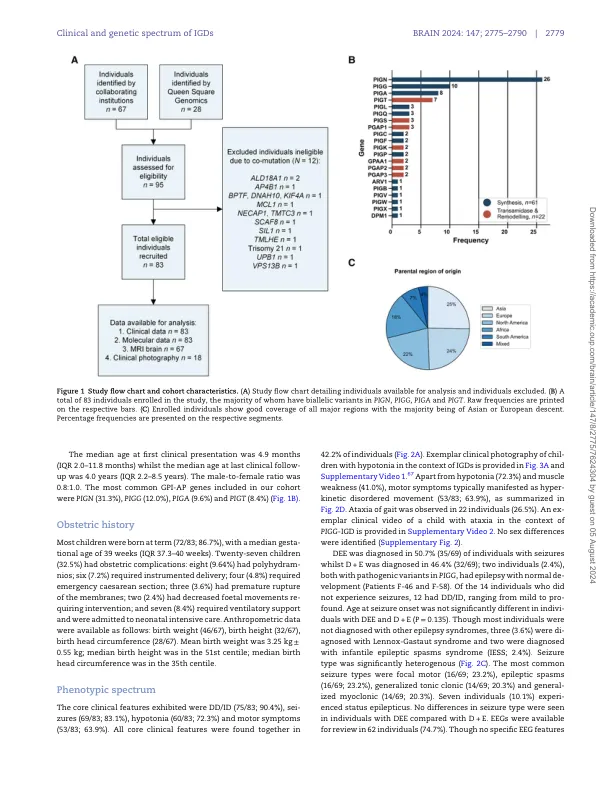
遗传性糖基磷脂酰肌醇缺乏症 (IGD) 是一组罕见的多系统疾病,由糖基磷脂酰肌醇锚定通路 (GPI-AP) 基因的致病变异引起。尽管至少 31 个 GPI-AP 基因中有 24 个与人类神经遗传疾病相关,但先前的报道仅限于单个基因,未考虑 GPI-AP 的整体情况,且自然史数据有限。在这项跨国回顾性观察研究中,我们系统分析了来自 75 个独特家族的 83 名 IGD 患者的分子谱、表型特征和自然史,其中包括 70 名新报告的个体;这是迄今为止最大的单一队列。核心临床特征是发育迟缓或智力障碍(DD/ID,90%)、癫痫发作(83%)、肌张力低下(72%)和运动症状(64%)。预后和生物学意义显著的神经影像学特征包括脑萎缩(75%)、小脑萎缩(60%)、胼胝体异常(57%)和中央被盖束对称性弥散受限(60%)。61例患者有多系统受累,包括胃肠道(66%)、心脏(19%)和肾脏(14%)异常。虽然82%的患者出现畸形特征,但没有一种畸形特征的患病率超过30%,这表明存在显著的表型异质性。所有患者的随访数据均已获得,其中15例在撰写本文时已死亡。癫痫发作时的中位年龄为6个月。携带GPI-AP合成期基因变异的个体,其癫痫发作时间显著短于携带转酰胺酶和重塑期基因变异的个体(P = 0.046)。40例患者患有难治性癫痫。大多数患者(95%)出现言语迟缓或失语,以及运动迟缓(需非救护车辅助)
遗传性糖基磷脂酰肌醇缺乏症的临床和遗传谱
主要关键词




相关文件推荐