XiaoMi-AI文件搜索系统
World File Search SystemHEK293T
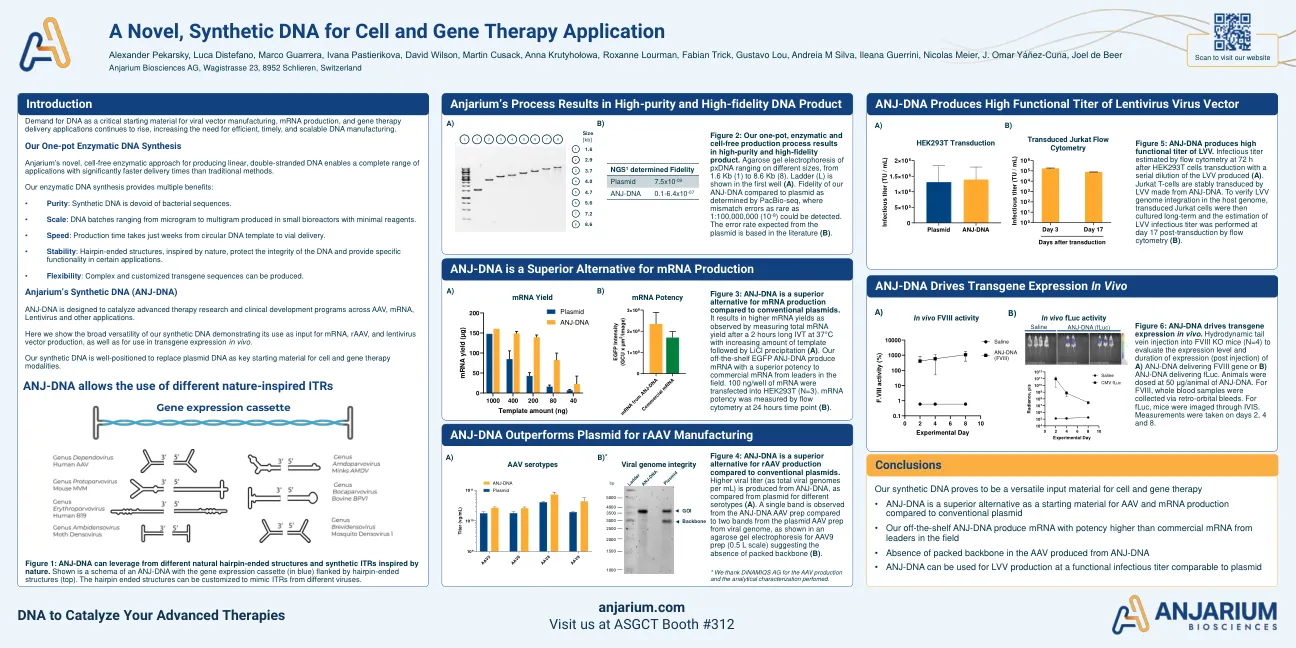
一种用于细胞和基因治疗应用的新型合成DNA
图3:与常规质粒相比,ANJ-DNA是mRNA产生的优越选择。它会导致较高的mRNA产量,如在37°C时测量2小时长的IVT后总我的mRNA产量,而模板量增加,然后是LICL沉淀(a)。我们现成的EGFP ANJ-DNA产生的mRNA具有较高的效力,可从该领域的领导者提供商业mRNA。mRNA的100 ng/孔被转染到HEK293T中(n = 3)。通过在24小时时间点(b)的流式细胞仪测量mRNA效能。

使用Cell -Profiler
是研究数字,维度,内容和分泌细胞器的定位的最常用和通用的方法之一是共聚焦显微镜分析。然而,可以在细胞中引起的分泌细胞器的数量,大小和形状中存在相当大的异质性。因此,需要分析大量细胞器以进行有效量化。正确评估这些参数需要一种自动,无偏的方法来处理和定量分析显微镜数据。在这里,我们描述了由Cell -Profiler软件运行的两个管道,称为OrganleleProfiler和OrganeLlecontentProfiler。这些管道线用于内皮菌落形成细胞(ECFC)的共聚焦图像,其中包含独特的分泌细胞器,称为Weibel-Palade体(WPB),以及ECFC和ECFC和人类胚胎肾脏293T(HEK293T)细胞的早期内体。结果表明,管道可以量化细胞计数,大小,细胞器计数,细胞器的大小,形状,与细胞和细胞的关系,以及在内皮和HEK293T细胞中与这些物体的距离。此外,使用管道来测量高尔基体破裂后WPB大小的减小,并在ECFC中触发CAMP介导的信号通路后量化WPB的核周聚类。此外,管道能够量化位于细胞器或细胞质中的二级信号,例如小的WPB GTPase RAB27A。使用斐济检查了细胞剖面测量值的有效性。确定,这些管道为多个细胞和细胞器类型的特性提供了强大的,高处理的定量工具。这些管道是免费的,可以在不同的细胞类型或细胞器上易于使用,并且易于编辑。

循环,与条件无关的活性在一级运动皮层中预测矫正运动行为
不同细胞群体的位点特异性遗传和表观遗传靶向是分子神经科学的核心目标,对于理解基因调节机制至关重要,这些基因调节机制是基于复杂的表型和行为的基础。虽然最近的技术进步已经实现了对基因表达的前所未有的控制,但其中许多方法都集中在选定的模型生物上和/或需要针对不同应用的劳动密集型定制。群集定期插入短质体重复序列(基于CRISPR)的系统的简单性和模块化已改变了基因组编辑并扩展了基因调节工具箱。但是,几乎没有可用于神经元细胞选择性CRISPR调节的工具。我们设计,验证和优化的CRISPR激活(CRISPRA)和CRISPR干扰(CRISPRI)系统用于CRE重组酶依赖性基因调节。出乎意料的是,基于传统的双流传式开放阅读框(DIO)策略的CRISPRA系统即使没有CRE也会显示出漏水的靶基因诱导。因此,我们开发了一种含有内含子的CRE依赖性CRISPRA系统(SVI-DIO-DCAS9-VPR),该系统减轻了泄漏基因诱导,并在HEK293T细胞和大鼠原发性神经元培养物中的内源基因上的传统DIO系统表现优于传统的DIO系统。使用基因特异性CRISPR SGRNA,我们证明了SVI-DIO-DCAS9-VPR可以以CRE特异性方式激活许多大鼠或人类基因(GRM2,TENT5B,FOS,SSTR2和GADD45B)。为了说明该工具的多功能性,我们创建了一个平行的CRISPRI构建体,该构建体仅在CRE存在下仅在HEK293T细胞中成功抑制了荧光素酶报告器的表达。这些结果为跨不同模型系统的CRE依赖性CRISPR-DCAS9方法提供了强大的框架,并在与常见的CRE驱动线或通过病毒载体交付时实现了细胞特异性靶向。
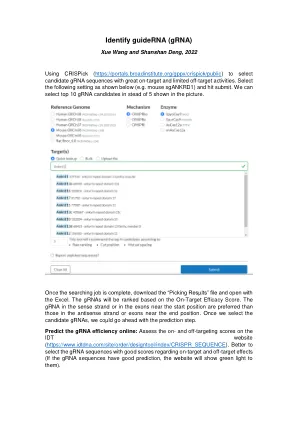
识别指南(GRNA)
第1天:转染。将GRNA - 黎病毒质粒与病毒包装质粒一起转染到HEK293T细胞中。第2天:第一批病毒的收获。早上用新鲜培养基代替转染介质,并在8小时后收集第一批细胞上清液。第3天:第二批和第三批病毒的收获。分别在清晨和午后收集细胞上清液。使用45μm的孔滤器过滤慢病毒上清液,以去除所有剩余的细胞碎片。立即使用它或等分病毒存储在-80°C下。3。病毒感染。

癌细胞国际诱导基因表达
图1创建合成cAMP响应元件结合蛋白(CREB)响应启动子。(a)腺苷信号传导的描述。腺苷(红色球)结合腺苷受体A2AR/A2BR,该腺苷受体动员相关的G蛋白(绿色)激活腺苷酸环化酶(橙色受体),并将ATP转化为3'5'- 5'-循环腺苷单磷酸腺苷(Camp)。另外,福斯科蛋白(橙色球)可以直接激活腺苷循环酶。CAMP结合蛋白激酶A(PKA)与磷酸化的CREB,该CREB结合了Plindromic DNA基序“ TGACGTCA”,激活了基因表达。(b)启动子设计和筛选示意图。cAMP响应元件基序(CRE,突出显示的黄色)被克隆在3倍重复中,两侧是鸟嘌呤“ G”(带下划线),六个散布的填充核苷酸(N)。3x Cres(灰色正方形)放在核心启动子(蓝色箭头)上游的1-6个重复中。用高斯荧光素酶(GLUC)或绿色荧光蛋白(EGFP)定量启动子活性。(c,d)HEK293T细胞在96个井板中用指示的构建体(x轴)反向转染。转染后48小时,用车辆(DMSO,浅蓝色条)或20μm福斯科林(FSK,深蓝色条)将细胞介质更改为培养基。八个小时后,对培养基进行了采样并测试了GLUC活性(RLU)。条表示n = 3实验重复的平均值,误差线代表标准误差(SEM)。**通过方差分析(ANOVA)Tukey检验,与所有其他样本相比,表示P <0.01。(E,F)流式细胞仪启动子诱导。HEK293T细胞用96个井板中的指定构建体(x轴)反向转染。转染后48小时,细胞培养基被更改为未处理的培养基(浅蓝色条),或补充了0.750 m m m腺苷(ADO,深蓝色条)的培养基。八个小时后,将细胞胰蛋白酶胰蛋白酶进行胰蛋白酶,并将其重悬于FACS缓冲液中以进行流式细胞仪。y轴表示正向散射(FSC)单元的EGFP中位荧光强度。条代表n = 3实验重复的平均值,误差线代表SEM。(g)启动子对腺苷的剂量反应性。HEK293T细胞在96个井板上反向转染,并在传说中指示的构造,然后培养48小时。然后更改培养基以添加不同的腺苷浓度,在8小时后进行采样,并测试了GLUC活性(RLU)。**通过12倍-CRE_YB的ANOVA TUKEY测试代表P <0.01,与1 m m的所有其他样品相比。每个点表示n = 3实验重复的平均值,误差线为SEM。

依赖 Cre 的 CRISPR/dCas9 系统用于调控神经元的基因表达
对不同细胞群体进行位点特异性遗传和表观遗传靶向是分子神经科学的核心目标,对于理解复杂表型和行为背后的基因调控机制至关重要。虽然最近的技术进步使对基因表达的控制达到了前所未有的程度,但其中许多方法都集中在选定的模型生物上和/或需要针对不同应用进行劳动密集型定制。基于 CRISPR 的系统的简单性和模块化已经改变了基因组编辑的这一方面,提供了各种可能的应用和目标。然而,目前很少有可用于神经元中细胞选择性 CRISPR 调控的工具。在这里,我们设计、验证和优化了 CRISPR 激活 (CRISPRa) 和 CRISPR 干扰 (CRISPRi) 系统,以实现 Cre 重组酶依赖性基因调控。出乎意料的是,基于传统双链倒置开放阅读框 (DIO) 策略的 CRISPRa 系统在没有 Cre 的情况下表现出泄漏的靶基因诱导。因此,我们开发了一种内含子 Cre 依赖性 CRISPRa 系统 (SVI-DIO-dCas9-VPR),该系统可缓解泄漏基因诱导,并且在 HEK293T 细胞和大鼠原代神经元培养物中,其在内源基因方面的表现均优于传统 DIO 系统。使用基因特异性 CRISPR sgRNA,我们证明 SVI-DIO-dCas9-VPR 可以以 Cre 特异性方式激活高度可诱导基因 (GRM2、Tent5b 和 Fos) 以及中等可诱导基因 (Sstr2 和 Gadd45b)。此外,为了说明此工具的多功能性,我们创建了一个平行的 CRISPRi 构建体,仅在存在 Cre 的情况下,它才成功抑制了 HEK293T 细胞中荧光素酶报告基因的表达。这些结果为不同模型系统中的 Cre 依赖性 CRISPR-dCas9 方法提供了一个强大的框架,并且当与常见的 Cre 驱动线或通过病毒载体进行 Cre 递送相结合时,将实现细胞特异性靶向。

残留宿主细胞 DNA 清除的评估和……
基于病毒载体的基因治疗药物产品可能含有来自生产细胞的残留 DNA。生产通常发生在具有致瘤潜力的连续细胞系中,残留宿主细胞 DNA 具有将致癌基因传播给受体细胞的理论风险。这种风险随着残留 DNA 片段浓度和大小的降低而降低。因此,监测基因治疗产品中残留宿主细胞 DNA 的浓度和大小非常重要。我们评估了在 HEK293T 细胞中制造的工程慢病毒载体生产过程中定量残留宿主细胞 DNA 浓度和大小的方法,并使用这些方法比较了两种市售核酸酶在下游加工步骤中的表现。

在体外评估聚乙胺的纳米插曲 -
有效的基因疗法依赖于有效的基因递送系统。病毒基因递送在转移和表达外部基因方面表现出色。但是,它们的免疫力和大规模生产的困难限制了其临床应用。相比之下,由于免疫原性较小,对大规模生产的便利性,基于纳米颗粒的基因递送系统的注意力越来越多。然而,与病毒系统相比,它们的转染效率差仍然是一个重要的障碍。在主题研究中,我们研究了在HEK293T,CALU-3,CALU-6细胞系和原代人骨髓间充质干细胞(MSC)中,我们调查了PEI涂层石墨烯氧化物的转染效率。氧化石墨烯的高表面比和良好的生物相容性使其成为基因递送系统的吸引力。但是,在水性环境中氧化石墨烯的低分散性是需要征服的第一个障碍。为此,我们通过在pH值为7的pH值中超声超声来增强水中氧化石墨烯在水中的分散性和稳定性。然后,将氧化石墨烯与分支PEI(25 kDa)偶联以具有局部电荷,从而使其能够将其凝结为具有天然负潜能的核酸。我们合成的纳米载体(GO-PEI)的生理化学特性由DLS,FT-IR和AFM确定。多聚体中使用的质粒包含GFP基因,从而使我们能够通过荧光显微镜和流式细胞体 - 尝试验证转染效率。虽然GO-PEI载体在转染HEK293T细胞方面高效,但MSC和Calu-3细胞的转移效率明显低。我们假设这些细胞中GO-PEI转染效率较低的主要原因是由于其较高的毒性。尽管如此,考虑到氧化石墨烯在药物输送中的各种优势以及其在生物医学中的光学和电气应用,我们建议用更具生物相容性材料功能化氧化氧化烯,以增强其作为这些细胞类型中基因载体的潜力。

预测基于 CRISPR-Cas9 的表观基因组编辑的效果
摘要表观遗传调控协调哺乳动物转录,但它们之间的功能联系仍然难以捉摸。为了解决这个问题,我们使用来自 13 种 ENCODE 细胞类型的表观基因组和转录组数据来训练机器学习模型,以预测组蛋白翻译后修饰 (PTM) 的基因表达,对于大多数细胞类型,实现了 ∼0.70 −0.79 的转录组范围相关性。我们的模型重现了组蛋白 PTM 和表达模式之间的已知关联,包括预测转录起始位点 (TSS) 附近的组蛋白亚基 H3 赖氨酸残基 27 (H3K27ac) 的乙酰化会显著提高表达水平。为了通过实验验证这一预测,并研究 H3K27ac 的天然沉积与人工沉积对表达的影响,我们将合成的 dCas9-p300 组蛋白乙酰转移酶系统应用于 HEK293T 细胞系中的 8 个基因和 K562 细胞系中的 5 个基因。此外,为了便于建立模型,我们执行 MNase-seq 来绘制 HEK293T 中全基因组核小体占有水平。我们观察到,我们的模型在准确排序基因对 dCas9-p300 系统的相对倍数变化方面表现良好;然而,与根据其天然表观遗传特征预测跨细胞类型的表达相比,它们对单个基因内倍数变化进行排序的能力明显减弱。我们的研究结果强调,我们需要更全面的基因组规模表观基因组编辑数据集,更好地理解表观基因组编辑工具所做的实际修改,以及改进因果模型,以便更好地从内源性细胞测量转移到扰动实验。这些改进将共同促进理解和可预测地控制动态人类表观基因组的能力,以及对人类健康的影响。

细菌回试启用人类细胞中的精确基因编辑
重试是参与抗流量防御的细菌遗传元素。它们具有将RNA转录为多拷贝单链DNA(MSDNA)的独特能力,该DNA保持与其模板RNA的共价链接。回合与酵母中的CRISPR-CAS9相结合,已显示可通过同源性定向修复(HDR)提高精确基因组编辑的编辑效率。HDR编辑效率受到与传递编码所需突变的细胞外供体DNA相关的挑战的限制。在这项研究中,我们测试了回发物作为供体DNA产生MSDNA的能力,并通过绑定MSDNA引导HEK293T和K562细胞中的RNA来促进HDR。通过使用CRISPR-CAS9系统的多个细菌物种的反性重构重构,我们证明了HDR速率高达11.3%。总的来说,我们的发现代表了将基于反性的精确基因编辑扩展到人类细胞的第一步。