机构名称:
¥ 1.0
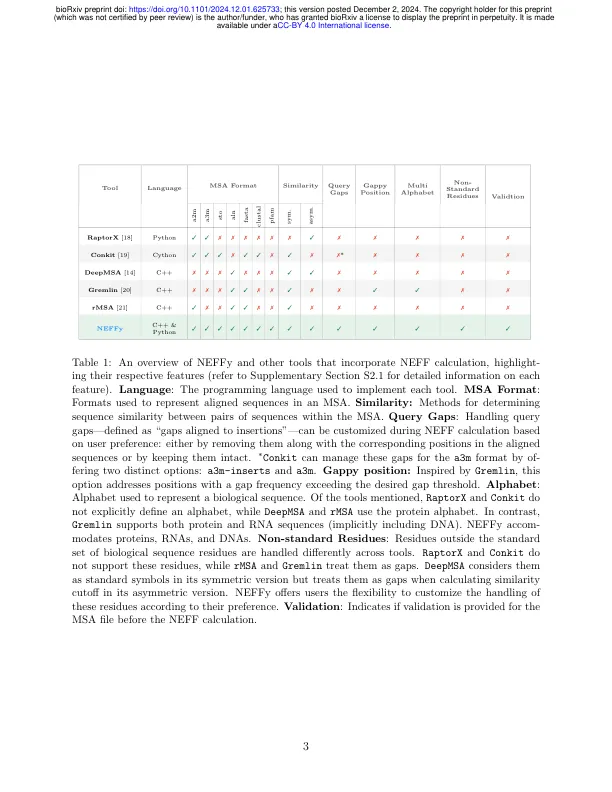
在最近的应用中,MSA的构建从有趣的查询顺序开始。该过程涉及搜索数据库以查找类似于查询的序列并将其对齐。DNA/RNA测序技术的最新进展扩大了Pub-LIC数据库,使能够产生具有高序列多样性的MSA [13,14]。通常认为这种MSA提供了更丰富的进化和协调性的见解,因此它们可以提高使用模型来下游任务的模型的有效性[9]。但是,由于MSA可以包含冗余序列,因此序列的数量本身可能不是其多样性的准确反映。“有效序列的数量”的概念,NEFF解决了这种冗余,并评估了MSA的质量。较高的NEFF值通常表明MSA更多样化和信息丰富,从而导致预测接触图和蛋白质或RNA分子的三级结构的精度[15,16]。例如,当NEFF值低于30 [5]时,Alphafold的准确性大大下降。此外,对于使用RNA的MSA作为输入的RNA结构预测模型(例如Trrosettarna),预测准确性与NEFF [7]相关,而对于高质量的MSA,这些模型可以胜过其他方法[17]。我们介绍了Neffy,这是一种快速而专用的独立工具,用于NEFF计算。neffy具有唯一装备的分析MSA,并在蛋白质和核酸序列的多种MSA格式中计算NEFF。它集成了NEFF工具(请参阅表1)中的所有功能,并提供一组新功能。neffy是在C ++中开发的,以实现最佳性能,并作为包装C ++可执行文件的Python库提供。这种方法可以使无缝集成到基于Python的工作流程中,从而简化了更广泛的受众的使用,同时保持效率。
neffy:用于计算有效序列数量的多功能工具
主要关键词




相关文件推荐