XiaoMi-AI文件搜索系统
World File Search SystemSMN2
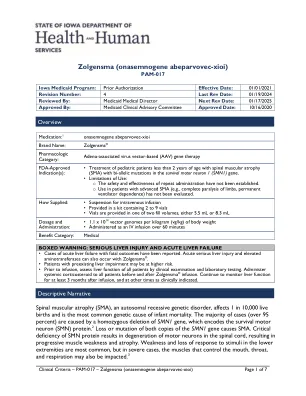
Zolgensma(onasengene abeparvovec-xioi)
结合SMN1的损失,患者保留了第二个相似基因SMN2的可变数量,这些副本可产生降低的生存运动神经元(SMN)蛋白水平,而这对于正常运动神经元功能不足。4 SMN2的副本数量较高,通常与温和的疾病相关,但是这种相关性是相对的,而不是绝对的相关性。5Zolgensma®(Onasengene Abeparvovec-XIOI)是一种基于AAV9的重组基因疗法,旨在提供编码人类SMN蛋白的基因的副本。在人类案例研究中,静脉内(IV)Zolgensma®的给药导致SMN蛋白的细胞转导和表达。脊柱肌肉萎缩已根据症状严重程度和基因型分类为0-4,但是有了新的疗法(包括Nusinersen,Risdiplam和Onasemnogene abeparvovec-Xioi),表格已经变得更加多样化和分类以集中在功能或治疗响应上。脊柱肌肉萎缩的分类6

5Q-SMA
摘要。目的:许多研究一直发现,SMN蛋白表达降低不会严重影响SMA患者的认知功能。但是,在不同的研究中,SMA患者的平均智能商在以上范围低于平均水平。通过新生儿筛查确定的SMA患者的认知发展仍然很大未知。方法:在2018年1月至2020年12月之间,使用Bayley III(BSID)在2岁之后,通过新生儿筛查确定的39个家庭中的47名合格SMA患者(23名女性/17名男性)中有40名。平均年龄为29.25个月(23-42个月)。17例患者患有2、11例患者有3例和12例患者的SMN2份≥4例。结果:认知量表:平均94.55(SD 24.01);语言量表:平均86.09(SD 26.41);运动量表:81.28(SD 28.07)。总体而言,认知量表表明,有14名儿童低于平均水平,20名儿童为平均水平,6名儿童高于平均水平。10/14分数低于平均水平的儿童有2份SMN2副本。事后成对比较表明,与BSID的电动机主尺度相比,认知主量表对SMN2副本的数量的敏感性要高(M = 10.27,p = 0.014)。也有证据表明,认知得分高于语言主量表(M = 7.11,p = 0.090)。结论:尽管早期开始治疗,但SMA儿童的认知儿童的认知发展受损,但强调了SMN蛋白在大脑发育的早期阶段的关键作用。

SALSA® 参考选择和分类 DNA SD084-S02
131 参考 00797-L25925 A1 2 136 参考 18457-L23634 A1 2 143 SMN1 / 内含子 7 S0938-L26163 A1 nag27134T>G 148 SMN1 / 外显子 8 S0961-L25586 A1 nag27706-27707delAT 154 SMN1 / 外显子 8 S0960-L25957 A1 2 163 参考 02291-L17086 A1 2 172 参考 02978-L17087 A1 2 183 SMN1 / 外显子 7 14919-L17081 A1 2 191 参考 00559-L17088 A1 2 200 参考 00976-L17298 A1 2 208 参考 12490-L17096 A1 2 228 参考 14498-L17101 A1 2 237 参考 02334-L17301 A1 2 245 参考 14293-L17100 A1 2 255 参考 13128-L17099 A1 2 264 参考 07630-L17091 A1 2 272 参考 14361-L17098 A1 2 282 SMN2 / 外显子 7 14921-L17083 A1 2 292 参考 18491-L23716 A1 2 301 参考 12783-L13918 A1 2 311 参考 06425-L17092 A1 2 321 参考 01042-L17093 A1 2 331 参考 01043-L17094 A1 2 注意:此处使用的突变命名法可能与文献不同!此 SD084-S02 参考选择和分箱 DNA 产品说明中使用的外显子编号是传统的外显子编号(外显子 1、2a、2b 和 3-8)。此外显子编号与 SMN1 和 SMN2 的 NCBI 参考序列不同。有关所用外显子编号和基因转录本的更多信息,请查阅相应的探针混合物产品说明。

对脊髓肌萎缩症患者体重≤17kg且≤24个月大的患者的骨nasegene abeparvovec的安全性和耐受性:
关键纳入标准•基于基因突变分析的症状SMA诊断,双重SMN1突变(缺失或点突变)和任何数量SMN2基因的副本•治疗时的年龄≤24个月•重量≤17kg筛选时访问时访问或批准的药物均可进行批准的药物,•纽约批准的药物/牢固的纽约,键/牢记的键abeparvovec在筛查前的四个星期内使用或先前使用任何AAV9基因疗法•抽吸史或吸气迹象(例如,食物的咳嗽或溅射)在筛查前的四个星期内在筛选前15天内

Nusinersen(Spinraza)
•神经科医生的处方•患者对5q-独生体隐性隐性脊柱肌肉萎缩(SMA)的诊断为SMN1基因中的双重缺失或突变•患者患者具有2至4份SMN2基因的副本•患者没有使用SMA治疗的治疗方法。 (ZOLGENSMA)或其他用于SMA的基因疗法•患者可以无永久的入侵通气或气管造口术呼吸*•患者每天醒来时可以呼吸而无需入侵或非侵入性通风,在每天醒来期间,患者•KAISER永久性咨询的持续咨询及其在使用中,并在进行持续的医疗疗法(继续使用)。治疗期间每12个月进行一次审查)。在以下情况下建议中断:

特异性,协同性和剪接修改药物的机制
Title: Specificity, synergy, and mechanisms of splice-modifying drugs Authors : Yuma Ishigami 1,† , Mandy S. Wong 1,2,† , Carlos Martí-Gómez 1 , Andalus Ayaz 1 , Mahdi Kooshkbaghi 1 , Sonya Hanson 3 , David M. McCandlish 1 , Adrian R. Krainer 1,* , Justin B. Kinney 1,*。隶属关系:1。Cold Spring Harbour实验室,纽约州冷泉港,美国11724,美国。2。当前地址:横梁治疗学,马萨诸塞州剑桥,美国02142,美国。3。flatiron Institute,纽约,纽约,10010,美国。†同等贡献。*通信:krainer@cshl.edu(ark),jkinney@cshl.edu(jbk)。摘要:针对MRNA剪接的药物具有很大的治疗潜力,但是对这些药物的工作原理的定量了解受到限制。在这里,我们引入了机械解释的定量模型,以针对剪接修改药物的序列特异性和浓度依赖性行为。使用大量平行的剪接测定,RNA-seq实验和精确剂量反应曲线,我们获得了两种用于治疗脊柱肌萎缩的两种小分子药物Risdiplam和Branaplam的定量模型。的结果定量地表征了Risdiplam和Branaplam对于5'剪接位点序列的特异性,这表明Branaplam通过两种不同的相互作用模式识别5'剪接位点,并证明了SMN2 Exon 7的Risdiplam活性的普遍的两点假设。结果还表明,在小分子药物和反义寡核苷酸药物中,异常的单药合作以及多药协同作用是促进外生包容的。Nusinersen 11–我们的定量模型阐明了现有治疗的机制,并为新疗法的合理发展提供了基础。引言替代性mRNA剪接已成为药物发育的主要重点1-10。已经开发了三种剪接改良药物 - Nusinersen,Risdiplam和Branaplam,以治疗脊柱肌肉萎缩(尽管Branaplam已撤回)。所有三种药物都通过促进SMN2外显子7。

遗传性神经肌肉疾病的基因靶向治疗
Nusinersen 是一种反义寡核苷酸 (ASO),可结合 SMN2 前信使核糖核酸 (RNA),8 通过增加外显子 7 的包含来稳定转录,从而增加功能性 SMN 蛋白质的产生。Nusinersen 以 2 个月内 4 次负荷剂量鞘内给药,随后在患者一生中每 4 个月维持一次剂量。罕见的副作用包括血小板减少、凝血异常和肾毒性,每次给药时都会监测所有这些副作用。8 如果发生严重的脊柱侧弯,尤其是既往脊柱融合,那么在以后的生活中接受治疗的较轻 SMA 患者鞘内入路可能会很困难。随着时间的推移,腰椎穿刺相关并发症以及需要镇静的患者反复接触镇静剂会影响耐受性。

Spinraza(Nusinersen)
脊柱肌肉萎缩(SMA)是一种常染色体隐性遗传疾病2,影响10,000例活产的1分之一,并且是婴儿死亡率的最常见遗传原因。大多数病例是由位于5q13处的生存运动神经元1(SMN1)基因中的突变引起的(95%的SMN1外显子7或从SMN1到SMN2的基因转换的纯合缺失)。这些突变会导致运动神经元的进行性变性,从而导致肌肉萎缩。下肢中对刺激的无力和对刺激的反应丧失是最常见的,但是在严重的情况下,控制口腔,喉咙和呼吸的肌肉也可能受到影响。医疗保健专注于呼吸支持,营养支持,抗生素呼吸道感染的管理以及通过支撑,物理治疗和手术的肌腱染色体和脊柱侧弯的管理。尽管最多的寿命

Bio-Rad QXDX AUTODG DDPCR系统脊柱肌肉萎缩指南
图2-诊断算法:在患者中对SMA的临床怀疑后,使用MLPA或QPCR对SMN1缺失进行诊断测试,以及同时对SMN2进行同时测试,可以迅速,准确地分类SMA并选择治疗。如果未检测到SMN1的副本,它确认了SMA的诊断。如果找到了一个SMN1的副本,则需要对SMN1基因进行进一步的测序,以确定检测到的副本是否包含突变。单个SMN1副本中的一个突变证实了SMA的诊断,而没有突变表示其他形式的SMA或NMD的可能性。在检测到两个或更多份SMN1的情况下,研究了患者的血缘家族病史。如果存在血父,则进行进一步的测序以评估潜在突变。相反,缺乏血缘关系表明其他SMA或NMD疾病的可能性。3.4.7。如果临床

Swapnil Tirmanwar,Int。 J. of Pharm。 Sci。,2025,第3卷,问题01,131-136
脊柱肌肉萎缩(SMA)是一种主要影响运动神经元的遗传神经肌肉疾病,导致渐进的肌肉无力和萎缩。它是由SMN1基因突变引起的,这导致生存运动神经元(SMN)蛋白的水平降低,这对于肌肉功能必不可少。SMA以不同类型的形式呈现,从严重的婴儿发作形式到较轻的成人发作病例,以及影响流动性,呼吸和运动发育的症状。此临床评论概述了SMA的遗传基础,分类,症状,症状,症状和诊断方法。它还研究了治疗策略的进步,包括基因疗法(例如Onasemnogene Abeparvovec),SMN2剪接修饰剂(如Nusinersen)以及支持护理以改善生活质量。该评论进一步强调了早期诊断,疗法的可及性以及新生儿筛查计划对更好结果的重要性。目标是对SMA进行全面的了解,以告知临床医生,看护人和公共卫生从业人员有效的疾病管理和新兴治疗选择。