
机构名称:
¥ 1.0
人们对二维过渡金属二硫属化物产生了浓厚的兴趣,这引发了大量使用可扩展气相方法(如化学气相沉积 (CVD) 和原子层沉积 (ALD))对其合成进行实验研究。ALD 通常允许较低的沉积温度,并且化学前体的成核需要与表面功能团发生反应。研究 ALD 建模的常用第一性原理方法是计算拟议反应途径的活化能。在这项工作中,我们使用密度泛函理论 (DFT) 计算了部分电荷密度、局部态密度 (LDoS)、Bader 电荷分析、吸附能和电荷密度差,以研究 MoF 6 在三种氧化物表面(包括 Al 2 O 3 、HfO 2 和 MgO)的成核。我们的研究结果表明,羟基 (OH) 有助于降低 MoF 6 前半周期内的反应势垒并促进前体在氧化物基底上的化学吸附。这一发现得到了氧化物表面高离子性 MF x(M = 金属,x = 1、2、3)键形成的支持。通过比较有羟基和无羟基的表面,我们强调了表面化学的重要性。
MoF6 在羟基化催化剂上吸附的第一性原理研究...
主要关键词



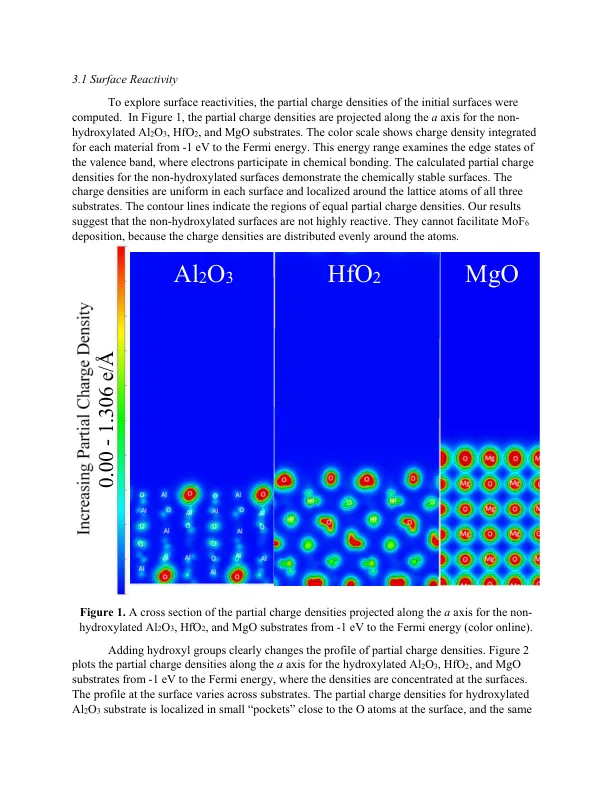
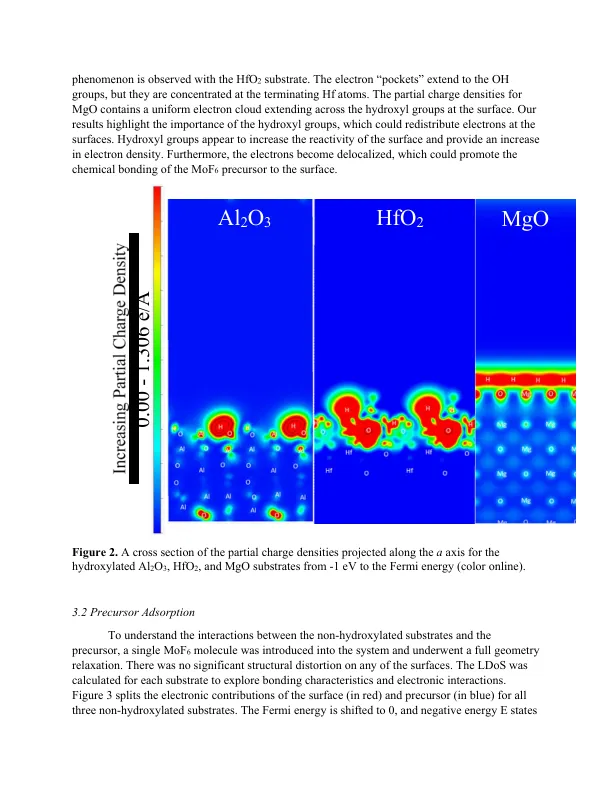
相关文件推荐





























