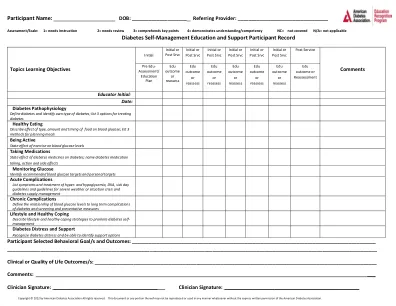
机构名称:
¥ 2.0
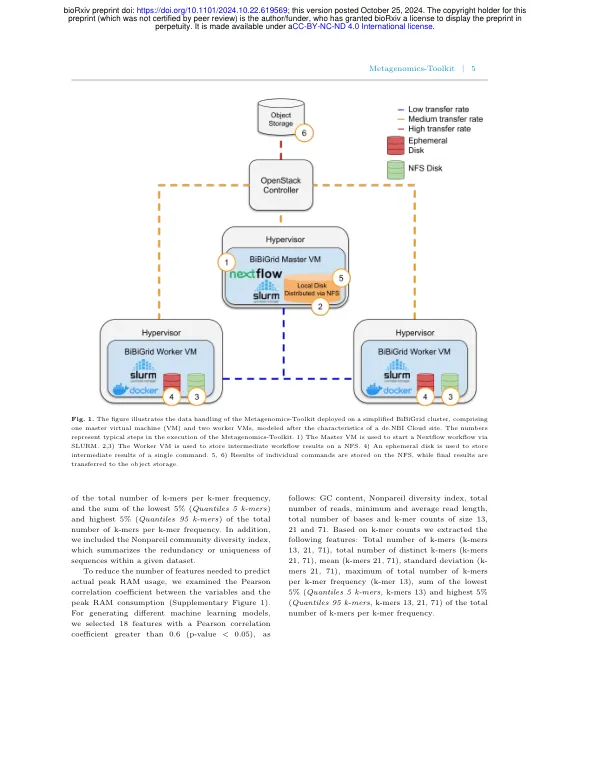
对具有数千个数据集的复杂环境的元基因组分析,例如序列读取存档中可用的数据集,需要巨大的计算资源才能在可接受的时间范围内完成计算工作。这样的大规模分析要求有效地使用基础基础结构。此外,任何分析都应完全可复制,并且必须公开使用工作流程,以允许其他研究人员了解计算结果背后的推理。在这里,我们介绍了可扩展的数据不可吻合的工作流程的宏基因组学-Toolkit,该工作流程分别自动化了从Illumina或Oxford Nanopore技术设备获得的简短和长元基因组读取。宏基因组学-ToolKit不仅提供元基因组工作流程中预期的标准功能,例如质量控制,组装,套件和注释,还提供独特的特征,例如基于各种工具的质粒识别,恢复无组成的微生物社区成员的恢复以及通过互联网模型的相互依赖模型的恢复,并发现了一个元素,并依赖于基本模型,并进行互联网组合。此外,元基因组学 - 库尔基特包括一个机器学习优化的组装步骤,该步骤量身定制元素组装程序要求的峰值RAM值以符合实际要求,从而最大程度地减少对专用高音硬件的依赖性。虽然可以在用户工作站上执行Metagenomics-Toolkit,但它还为有效的基于云的集群执行提供了多种优化。Metagenomics-Toolkit是开源的,可在https://github.com/metagenomics/metagenomics-tk上获得。我们将宏基因组学 - toolkit与五个常用的宏基因组工作流进行了比较,并通过在757 Metagenome数据集中从污水处理样本中执行元基因组学 - 静电库来证明其能力,以研究可能的污水核心微生物组。
宏基因组学-Toolkit
主要关键词




相关文件推荐