
机构名称:
¥ 2.0
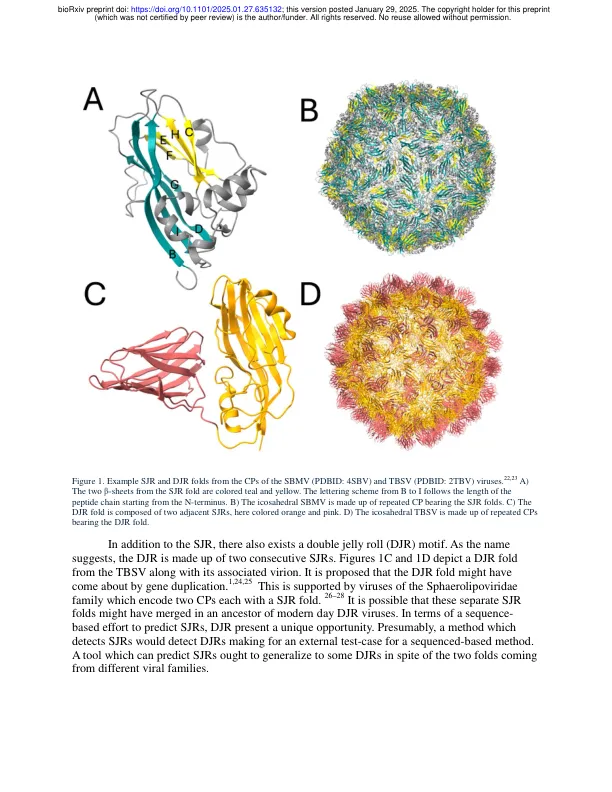
jrseek:人工智能在病毒中遇到果冻卷折叠分类,杰森·E·桑切斯(Jason E. Sanchez)1,温汉·朱2(Wenhan Guo 2),丘奇安格李3,林李3 *,chuan xiao 2 * 1计算科学系,德克萨斯大学El Paso,El Paso,El Paso,El Paso,TX 2德克萨斯大学埃尔帕索分校的物理学,德克萨斯州埃尔帕索 *通信:电子邮件:lli5@utep.edu; cxiao@utep.edu关键字病毒;人工智能;机器学习;果冻卷;病毒结构摘要果冻卷(JR)折叠是病毒的衣壳和核蛋白质中发现的最常见的结构基序。其在许多不同病毒家族的动机中的普遍性开发了一种工具来预测其从序列中的存在。在当前的工作中,在六个不同的大语模型(LLM)嵌入训练的逻辑回归(LR)模型在将JR与非JR序列区分开时表现出超过95%的精度。用于训练和测试的数据集包括来自单个JR病毒,非JR病毒和非病毒免疫球蛋白样β-三明治(IGLBS)蛋白的序列,这些蛋白与JR结构上非常相似。鉴于病毒家族之间的低序列相似性和数据集的平衡性质,高精度尤其显着。同样,模型的准确性与LLM嵌入无关,这表明预测病毒JR折叠的峰精度更多地取决于数据质量和数量,而不是使用所使用的特定数学模型。鉴于许多病毒式衣壳和核素结构尚未解决,因此使用基于序列的LLMS是一种有前途的策略,可以轻松地应用于可用数据。Bert-U100嵌入的主成分分析表明,大多数IGLBS序列和JR和非JR序列的一个子集甚至在应用LR模型之前也可以区分,但是LR模型对于区分更歧义序列的子集是必要的。应用于双JR折叠时,BERT-U100模型能够为某些病毒家族分配JR图案,从而提供了该模型可推广性的证据。对于其他家庭而言,没有观察到这种概括性,激发了未来开发以双JR折叠告知的其他模型的需求。最后,BERT-U100模型还能够预测未分类病毒数据集中的序列是否产生JR倍数。给出了两个示例,JR预测由AlphaFold3证实。总的来说,这项工作表明JR折叠可以从其序列中预测。
JRSEEK:人工智能符合病毒中的果冻roll折叠分类
主要关键词




相关文件推荐





























