机构名称:
¥ 1.0
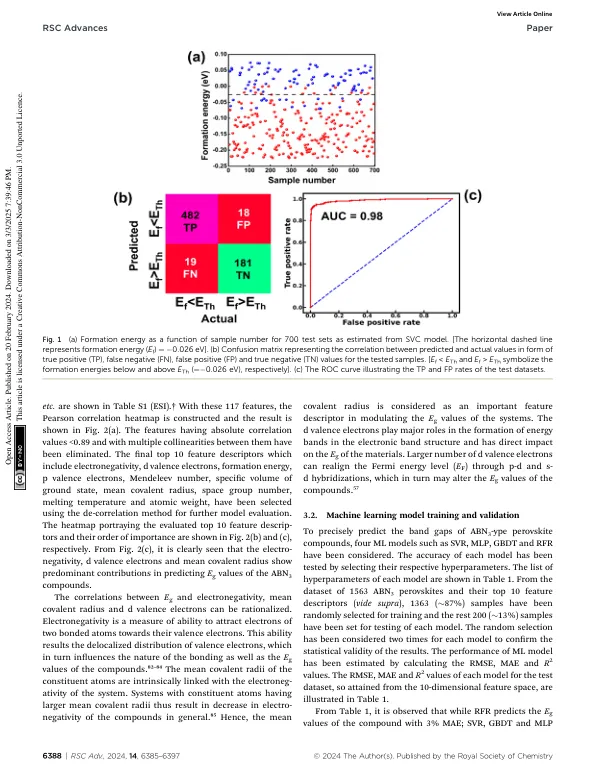
并提出极有可能通过实验实现。19 最近,人们利用第一性原理 DFT 计算来计算某些稀土氮化物钙钛矿 ABN 3(A = La、Ce、Pr、Nd、Pm、Sm、Eu、Gd、Tb、Dy、Ho、Er、Tm、Yb、Lu 和 B = Re、W)的磁矩和热力学稳定性,并提出了它们在氮化物材料领域的众多技术应用。16 在这方面,DFT 现在被认为是一种估算所研究材料的电子和光电特性的优雅方法。电子和光电特性主要由材料的带隙决定。虽然采用局部密度近似 (LDA) 和广义梯度近似 (GGA) 的 DFT 计算低估了 E g 值,33 – 36 但未经筛选的混合函数和 Perdew – Burke – Ernzerhof – Hartree – Fock 交换 (PBE0) 函数会高估化合物相对于其实验对应物的带隙能量。37 – 39 在这方面,使用混合交换关联 (XC) 函数,例如 Heyd – Scuseria – Ernzerhof (HSE)、Becke-3 参数-Lee-Yang-Parr (B3LYP) 和 B3PW91,通过单次 GW (G 0 W 0 ) 近似完成的 DFT 计算可以预测接近实验结果的化合物的 E g 值。 14,33,40 – 48 此类计算的主要缺陷在于它们对计算要求高并且需要高端服务器来运行它们。在这种情况下,机器学习(ML)现在被认为是一种有效的替代途径,可以避免与 DFT 计算相关的固有计算成本,并有助于在材料特性和目标变量(此处为 Eg)之间建立一个简单的模型。49 – 60 尽管最近已成功实施 ML 方法预测氧化物、卤化物钙钛矿和双钙钛矿化合物的带隙,61 – 66 但在预测氮化物钙钛矿的带隙方面尚未发现此类报道。考虑到上述问题,本文旨在从 ML 模型中预测 ABN 3 钙钛矿的带隙。已经进行了 DFT 研究以估计两种新型氮化物钙钛矿 CeBN 3(B = Mo,W)的电子能带结构、Eg 值和光电特性。本文的结构如下:第2节讨论了计算方法,包括ML方法和第一性原理DFT计算。第3.1节分享了ABN 3钙钛矿数据的清理和预处理。第3.2节讨论了ML模型的训练和验证。第3.3节致力于理解两种新发现的氮化物钙钛矿化合物CeBN 3 (B = Mo, W)的结构性质和稳定性。第3节。图4以CeBN 3 化合物的电子能带结构和带隙计算为框架,采用不同层次的DFT理论进行计算。相应的光电特性已在第3.5节中重点介绍。本研究的总体结论已在第4节中讨论。
预测 ABN3 钙钛矿的带隙 - RSC 出版社
主要关键词




相关文件推荐