机构名称:
¥ 1.0
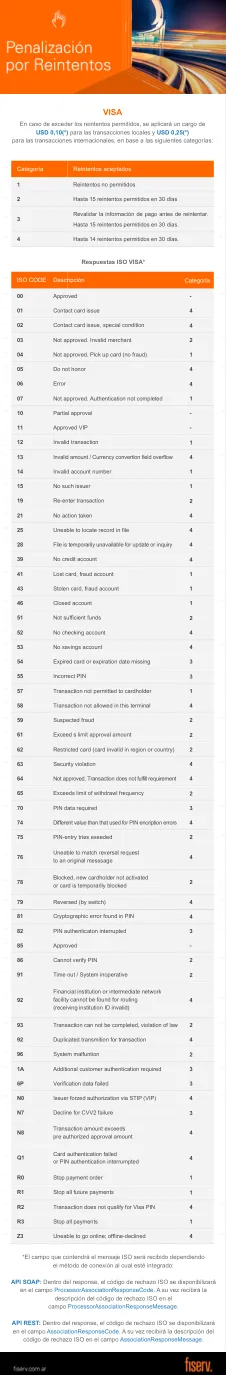
方法:从UCSC Xena和基因表达综合(GEO)数据库中提取了与CC和其他常见妇科癌有关的转录组数据和临床信息。在这项研究中,获得了CC(靶基因)的差异表达的CRRG,并通过“ clusterproFiller”进行了这些靶基因的功能富集分析。然后,将CC的生物标志物筛选为构建生存风险模型(风险评分)。此外,在不同风险组中进行了不同风险组的功能和肿瘤微环境(TME)分析,以进一步研究CC的潜在机制。此外,还进行了三种常见的妇科癌症中生物标志物的预后价值和功能分析,以揭示潜在的一致性或异质性法规。
转录组分析显示多个昼夜节律
主要关键词





相关文件推荐