机构名称:
¥ 1.0
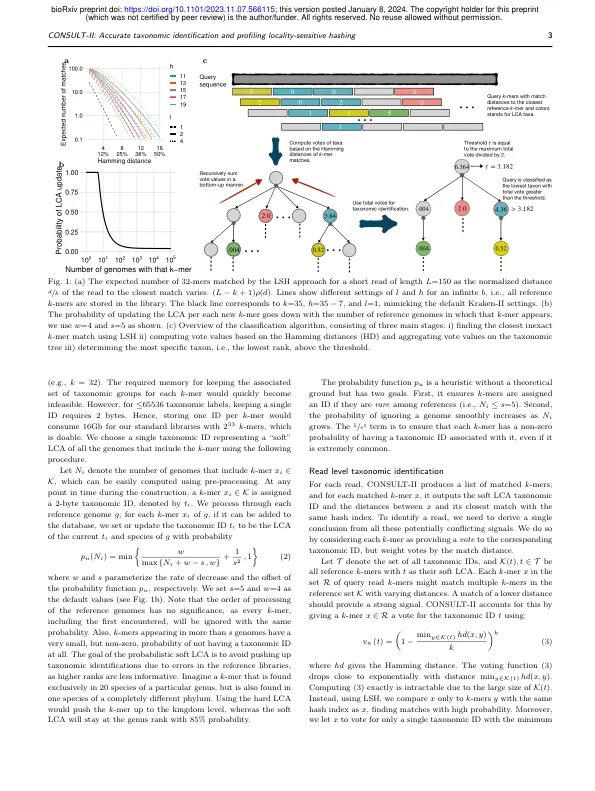
微生物群落的宏基因组测序产生了来自未知的微生物的简短DNA读数(Handelsman,2004),导致需要基于参考数据集的分类学识别。一种方法是从分类学上识别读取并总结结果以获得样本的分类学概况,显示了分类群体的相对丰度。但是,尽管有成熟的读取分类和分析工具的可用性,但基准测试揭示了现有方法的准确性的主要差距(McIntyre等人。,2017年; Meyer等人。,2019年; Sczyrba等。,2017年; Ye等。,2019年)。精确的识别通常会受到查询的新颖性与全基因组参考数据集和模棱两可的匹配的阻碍。此外,对大量基因组进行搜索是计算要求的。分类学识别方法采用各种策略,包括K -Mer匹配(Ames等人,2013年; Ounit等。,2015年;伍德等。,2019年; Lau等。,2019年; Lu等。,2017年),阅读映射(Zhu等人,2022),基于标记的对准(Liu等人。,2011年;米兰等。,2019年; Segata等。,2012年; Sunagawa等。,2013年)和系统发育放置(Asnicar等人。,2020年; Shah等。,2021; Truong等。,2015年)。无论如何,它们本质上都搜索了样本中的读数和参考集之间的匹配。,2017年),尤其是在众所周知的微生物栖息地(如海水或土壤)中(Pachiadaki等人。,2019年)。挑战是地球微生物多样性的很大一部分缺乏参考数据集中的近距离代表(Choi等人因此,大多数方法
咨询-II:使用对区域敏感的哈希
主要关键词




相关文件推荐