XiaoMi-AI文件搜索系统
World File Search SystemSAES

ondexxya®(andexanet alfa)
在合并的OnDexXYA(所有剂量)分析集中总共包括417名健康受试者,并将156名受试者包括在合并的安慰剂分析集中。仅接受推注剂量(从90毫克到800 mg)和274剂接受甘露剂量(从400至800 mg),然后连续输注4 mg/min(低剂量)或8 min min(高剂量)或120分钟(高剂量),持续120分钟(480毫克)(480 mg)。不管与治疗的关系如何(TEAES),经过46.8%的ONDEXXYA治疗受试者,而接受安慰剂的受试者中有43.6%的人经历了46.8%的治疗。一个严重的不良事件(SAE)(SAE)(肾结石病)发生在经过ONDEXXYA治疗的健康志愿者中,安慰剂组中没有发生SAE。两个经过ONDEXXYA治疗的健康受试者有TEAE(输注相关反应),导致该研究过早停用。在安慰剂组中未观察到该研究的过早中断。表4中显示了合并健康志愿者研究中最常见的茶(见8.2临床试验不良反应)。

包装说明书 - AUDENZ
AUDENZ 在成人(18 岁及以上)中的临床安全性数据来自三项研究:研究 1 针对 18 至 64 岁的成人(NCT01776541);研究 2 针对 65 岁及以上的成人(NCT01766921),研究 3 是针对 18 岁及以上成人的安慰剂对照试验(NCT02839330)。所有研究中的受试者均接受了 2 剂 AUDENZ,肌肉注射间隔 21 天。在所有三项研究中,收集了每次接种后 7 天内征集的局部和全身不良反应,并收集了每次接种后 21 天内未征集的不良事件。每位受试者最后一次接种疫苗后一年内,收集了严重不良事件 (SAE)、特别关注的不良事件 (AESI)(代表潜在免疫介导疾病的前瞻性定义事件)、新发慢性病 (NOCD)(导致新诊断慢性疾病的不良事件)和就医不良事件 (MAAE)(导致非计划医疗就诊)。安全人群包括 3,579 名接受至少一剂 AUDENZ 的受试者。其中,1,683 人为 18 至 64 岁的成年人,1,896 人为 65 岁及以上的成年人。
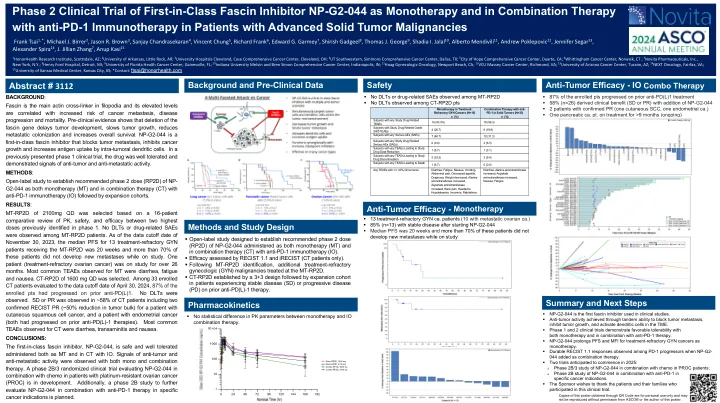
第一类Fascin抑制剂NP- ...
根据PK的16名患者比较审查,选择了2100mg QD的MT-RP2D,安全性,安全性和疗效。未观察到DLT或与药物有关的SAE。截至2023年11月30日的数据截止日期,接受MT-RP2D的13例治疗治疗症患者的中位PFS为20周,其中70%以上的患者在研究期间没有发展新的转移。一名患者(治疗难治性卵巢癌)正在研究超过26个月。观察到的MT最常见的茶是腹泻,疲劳和恶心。CT-RP2D为1600 mg QD。在2024年4月30日的数据截止日期评估的33名注册的CT患者中,有87%的招募PTS在先前的抗PD(L)1中进行了进展。未观察到DLT。SD或PR,包括两名患有皮肤鳞状细胞癌患者的患者,包括两名确认的RECIST PR(肿瘤大量降低50%)和子宫内膜癌患者(两者都在先前的抗PD(L)-1疗法上进展。CT观察到的最常见的茶是腹泻,跨动脉炎和恶心。

舌下 - 免疫疗法-AS-A-A-A-A-A-Technique-of-Alergen- ...
2020 Cochrane系统审查(SR)评估了舌下免疫疗法(SLIT)的功效和安全性,而成人和哮喘儿童的安慰剂或标准护理。[1]包括任何评估舌下免疫疗法和安慰剂或作为标准哮喘管理的辅助的任何盲目或任何持续时间的平行随机对照试验(RCT)。主要结果是哮喘恶化,需要访问急诊室(ED)或入院,验证的生活质量措施以及全因严重的不良事件(SAE)。此外,分析了哮喘症状评分,需要全身性皮质类固醇的恶化,对挑衅测试的反应以及吸入的皮质类固醇(ICS)剂量的次要结果。66项研究包括在审查中,主要由双盲和安慰剂对照试验组成,这些试验的持续时间从一天到三年不同。参与者患有轻度或间歇性哮喘,通常患有合并症过敏性鼻炎。数据无法从16项研究中进行分析,另外6项研究只能包括在所有不良事件的事后分析中。在大约四分之一的研究中存在高表现或检测偏差(或两者)的高风险,而在大约一半的研究中,损耗很高或未知。在大多数研究中没有报告审查中针对的主要结果;只有两项小型研究报告说需要急诊或医院就诊。这些研究的汇总估计

DM_in_Clinical Pharmacology.pdf
• 根据个人因素合理选择药物并调整给药方案。 • 对涉及药品的临床实践进行审计,并向临床医生提供适当的反馈。 • 制定和评估处方政策、基本药物清单、处方集和标准治疗指南 • 成为团队的一员,设计医院抗生素政策和抗生素管理活动。 • 成为团队的一员,向处方委员会和监管机构提交有关新旧药物纳入/排除的有效意见。 • 设计和开展人类志愿者药代动力学-药效学 (PK-PD) 研究和人群 PK-PD 研究。 • 设计和开展/或成为团队的一员,开展研究者发起的药物试验。 • 参与各种临床试验活动,如受试者招募、知情同意管理、随机化和盲法、试验受试者的临床评估、试验行为监测、严重药物不良反应报告。 • 参与严重不良事件 (SAE)、药物不良反应、药物间反应和用药错误的检测、报告、管理,包括因果关系评估(并向主管教授报告)。 • 参与其他药物警戒活动,如因果关系评估。 • 监测药物不良反应并向主管部门报告以进行管理。 • 应能够管理或提供有关药物过敏症的建议。 • 成为医院药物和治疗委员会以及其他相关医院委员会的一部分或参与其活动。

OBX-115在具有免疫检查点抑制剂(ICI)耐药性或转移性疾病>
†主要终点•安全性:不良事件(AES)和严重AES(SAE)的发生率和严重性•耐受性:剂量中断,减少和中断•识别剂量的识别:剂量限制毒性(DLTS)关键终点的发病率和性质•效率•效率:调查的效率为11剂量升级•同时1(n = 3) - OBX-115 30×10 9最大细胞(患者1接收了150×10 9个细胞和ACZ 500 mg/day,该协议的每日版本的每日版本) - ACZ 125 mg/天,从第2天到第9天到第9天到第9天到9•群体2B(N = 3-6)的2B(N = 3-6)的最大值(N = 3-6)and 2 cyl 2 cell 2 to Acd and Ac Ac/aw Nay to Acc/aw Nay 2 Acc/Ac ac ac ac ac ac ac c 125 c.11 ac c 12 5 mim c。将根据需要进行其他升级/降级队列的群体,关键资格标准•病理确认的转移性黑色素瘤的诊断(不可切除的III期或IV期)•复发和/或对ICI治疗的复发和/或对ICI疗法的复发和/或耐火性,包括抗或没有抗cttla-ctla-4 anti-anti-andibir of anti-ctla-4 anti-anti-antiby obx-les of-les of-les-les-les ofbody•肿瘤组织采购(TTP)的≥1病变(TTP)进行恢复v1.1反应评估•ECOG PS 0-1治疗方案•使用患者自身的肿瘤组织制造新鲜(非晶状体保存)OBX-115资格)用环磷酰胺和氟达拉滨(图2)•ACZ每天以同类剂量口服一次,每天一次服用7天,在第6-8周,如果初始肿瘤响应不足,则在第6-8周进行额外的ACZ,如果不局部反应不足(PR)•没有系统性的高剂量IL2是全身性高剂量IL2,则是全身性数据截止日期:1月2日,2024年1月2日,20224

国防部自动测试系统总体规划 - Osd.mil
1.0 执行摘要 本文档为国防部 (DoD) 自动测试系统 (ATS) 采购政策和投资战略的实施提供了综合总体规划。它研究了国防部 ATS 采购管理政策的历史演变,描述了各军种的 ATS 管理组织,确定了国防部 ATS 管理结构的主要参与者,并定义了不断发展的国防部 ATS 现代化战略。该计划详细说明了实施参考文献 (a)、(b) 和 (c) 中所述的国防部 ATS 采购政策所涉及的管理流程,该政策指导各军种通过使用指定的 ATS 系列作为首选解决方案来满足自动测试设备 (ATE) 硬件和软件需求,以最大限度地降低总生命周期拥有成本。ATS 总体规划提出了指定未来国防部 ATS 系列和向当前指定系列添加测试仪的既定标准。它讨论了获得批准采购不符合国防部 ATS 政策的自动测试仪的过程。它引用了使用参考 (d) DoD ATS 选择流程选择和实施 ATS 解决方案以满足武器系统要求所需的工具。ATS 总体规划是根据服务采购主管 (SAE) 之间的协议发布的,如联合协议备忘录 (参考 (e) 中所述。DoD ATS 执行局 (ATS ED) 负责定期审查和更新 DoD ATS 总体规划。2017 年 DoD ATS 总体规划取代了 2012 年及之前的 DoD ATS 总体规划。

2024年11月13日,亲爱的右病人倡导领袖,...
•高剂量的TSHA-102通常耐受性良好。TSHA-102通常在前两个青少年/成年患者中,最多20周,在最高六个星期的第一批儿科患者中,在前两个青少年/成年患者中,没有严重的不良事件(SAE)或剂量限制性毒性(DLT)。对第三个高剂量的成人/青春期患者进行了给药,并招募了第二个高剂量儿科患者。•持续入学高剂量队列。独立数据监测委员会(IDMC)审查了来自两个高剂量的成人/青少年参与者的可用临床数据,以及第一个高剂量的儿科参与者,并批准了在高剂量水平的青少年/成人和儿科研究中招收的程序。•完成了阳性再生医学高级治疗(RMAT)B型会议。与食品药品监督管理局(FDA)一致,在该机构对可用数据的审查之后,关于TSHA-102的持续开发方法以及B部分试验设计,端点以及既定自然历史数据集的潜在使用。•提出的生物分布数据进一步支持鞘内递送的临床潜力。在2024年10月在欧洲欧洲基因和细胞疗法欧洲欧洲基因和细胞治疗学会的第31届年度大会上,介绍了评估AAV9基因治疗递送的五项非人类灵长类动物(NHP)研究的分析的数据。请在下面找到一些常见问题的答案列表。揭示青少年和成人(12岁以上)的目标是什么?

BEACON-NEUROBLASTOMA协议_VN 8.0_VD 07MAR2023
删除了传真紧急随机化的选项。仅在第6.2节中对第7.2节中的指南进行更改,以计算体重超过年龄第98世纪的患者的剂量计算。在第7.6和9.1.2节中删除增强的数据收集,以进行特殊关注的不良事件(AESI)。在第7.6.1节中加入赞助商的一致性延长治疗延迟的选择增加了坏死性筋膜炎,这是一个不良事件,需要在第7.6.4节中停用bevacizumab。对第7.10节的更改,涉及记录患者医疗笔记中伴随药物的记录和双膦酸盐的给药。关于实验室不良事件报告的第9.1节的更改。澄清不需要在第11节中不需要进一步后续访问的患者的后续表格完成安排。在8.2.4和8.3.3节中更改贝伐单抗和伊立替康的准备和分配准则。澄清替莫唑胺施用之前的禁食安排已添加到第8.4.3节中。在第9.1.3.1节中应在预期的SAR表格上报告的事件的更改。在第9.2.6节中,应向F.Hoffman-La Roche Ltd报告SAE的SAE,在第14.4节中增加了试验管理小组会议频率。在第13.4.2节中更改了伊立替康随机化的措辞。增加了表8、9、10和11的肝毒性剂量降低剂量和终止的指南。提到国家协调中心的参考已更改为全国共同负责人。对赞助商的引用已更改为协调赞助商。

I型干扰素对全身性红斑狼疮患者的Aarifrolumab封锁,在两个3期试验的基因表达和蛋白质组学分析中调节关键的免疫病理途径
抽象目标是评估单次验证骨髓间充质基质细胞(BM-MSC)与慢性下背部疼痛(LBP)患者中的假安慰剂的功效。方法参与者是在2018年4月至2022年12月之间的一项前瞻性,双盲,对照研究中随机分配的,以接受假注射或在验证内注射2000万个同种异体BM-MSC中。第一个共同主要终点是通过改善视觉模拟量表(VAS)至少20%和20 mm的响应者的速率,或者在基线和12个月之间的OSWESTRY残疾指数(ODI)为20%。二级结构共同主要终点是通过基线和第12个月之间的定量MRI T2测量的盘流体含量评估的。次要终点包括疼痛VAS,ODI,短形式(SF)-36和所有时间点上临床上最小的临床差异(1、3、6、12和24个月)。我们确定了与基线至6个月之间同种异细胞注入相关的免疫反应。记录了严重的不良事件(SAE)。结果114例被随机分组(n = 58,BM-MSC组; n = 56,假安慰剂组)。在12个月时,未达到主要结果(在安慰剂组中,BM-MSC组为69%; P = 0.77)。组在所有次要结果上没有差异。没有与干预有关的SAE。得出的结论虽然我们的研究并未结论证明同种异体BM-MSC对LBP的功效,但该程序是安全的。MSC治疗LBP的长期结局仍在研究中。试验登记编号Eudract 2017-002092-25/Clinicaltrials。Gov:NCT03737461。