机构名称:
¥ 1.0
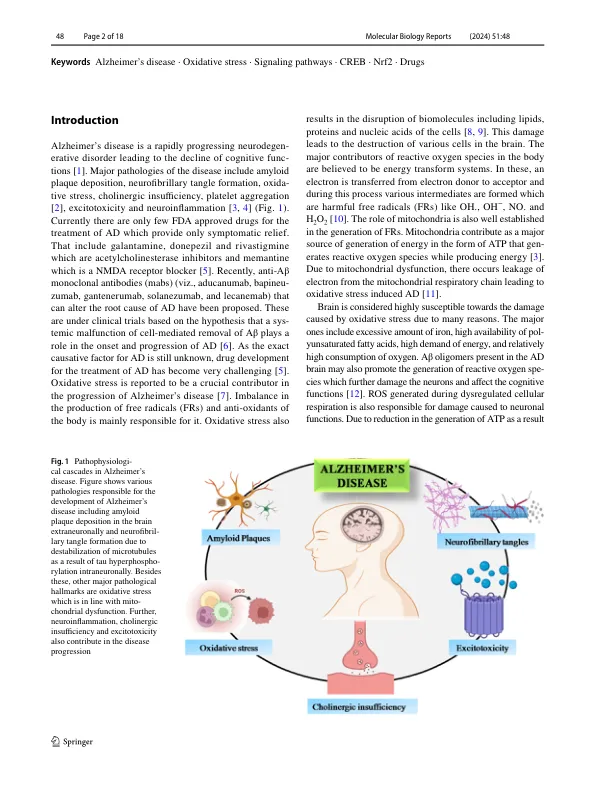
摘要阿尔茨海默氏病的病理生理学仍然是一个难题。越来越多的证据阐明了氧化应激参与AD的病理学,使其成为治疗性发育的主要靶标。由线粒体功能改变,电子传输链失调和其他来源产生的活性氧(ROS)提升了凝集的Aβ和神经原纤维缠结,从而进一步刺激了ROS的产生。氧化应激引起对脂质,蛋白质和DNA的损伤导致神经元死亡,从而导致AD。此外,氧化应激会诱导凋亡,这是由ERK1/2和NRF2途径的调节触发的,随后GSK-3β表达增加并降低了PP2A活性。氧化应激通过干扰RCAN1,CREB/ ERK,NRF2,PP2A,NFκB和PI3K/ AKT等各种信号通路来夸大疾病状况。研究报道了TNF-α在氧化应激刺激中的作用,该抗氧化剂刺激的作用增强了抗氧化剂水平。据报道,其他药物如普拉己烯,美金刚,卡维丝醇和褪黑激素可以激活CREB/RCAN1和NRF2途径。与此相一致,epigallocatechin Gallate和Genastein还靶向NRF2和CREB途径,从而导致下游途径(如AS和KEAP1)的激活,这些途径可以改善氧化应激条件。多奈酮和白藜芦醇减少氧化应激,并激活AMPK途径以及PP2A激活,从而促进tau去磷酸化和神经元存活。本研究详细描述了氧化应激在AD中的作用,涉及氧化应激诱导的AD的主要信号通路和正在针对这些途径的开发中的药物,这些途径可能有助于AD的治疗进展。
阿尔茨海默氏病的氧化应激
主要关键词




相关文件推荐