机构名称:
¥ 1.0
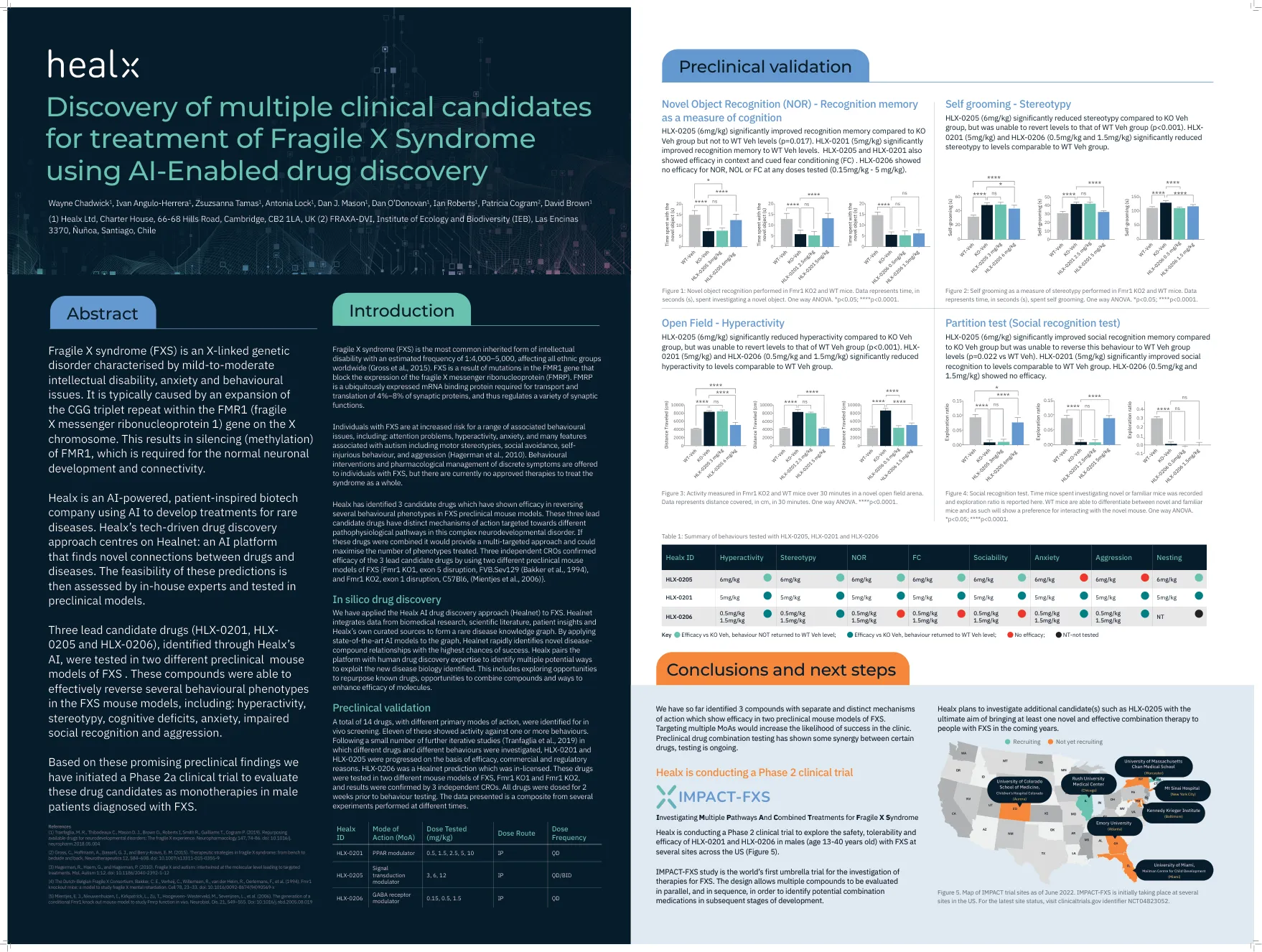
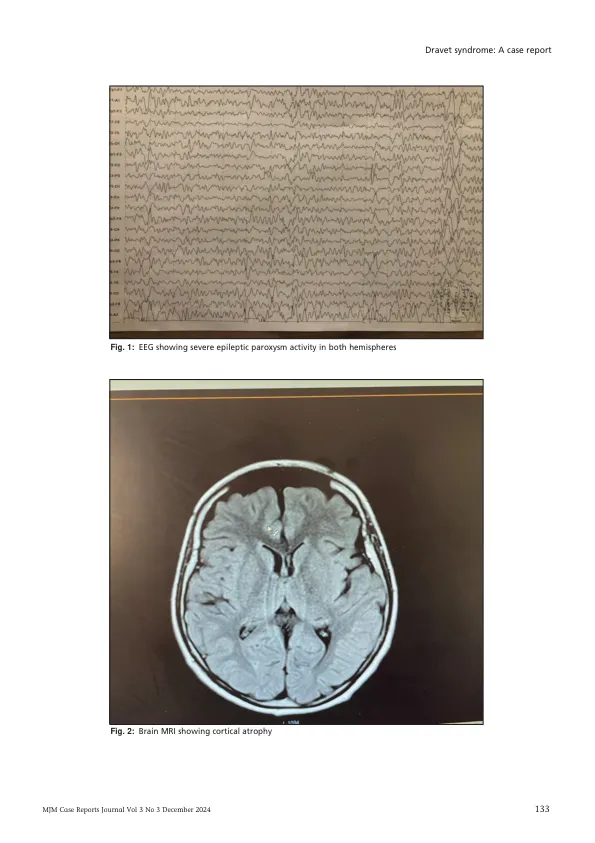
摘要 我们报告了一例罕见的 Dravet 综合征病例,患者是一名 13 岁男孩,因多形性多药耐药性癫痫发作而就诊。他出生后第一年反复发作几次热性惊厥,最初接受全身性癫痫治疗。但由于癫痫发作对一种抗癫痫药物有抵抗力,因此随着时间的推移,他增加了其他药物。全身性癫痫发作始于 8 个月大的热性惊厥,每次发热发作时经常重复发作,直到 5 岁时变为多形性,并出现认知和运动功能下降。没有相关家族史,也没有近亲关系。脑电图显示两个半球均有严重的癫痫放电,脑部 MRI 显示皮质萎缩。因此,该儿童被转诊进行 Dravet 综合征的基因检测,结果证实了 SCN1A 基因阳性突变的诊断。引言 Dravet 综合征 (DS) 以前称为婴儿严重肌阵挛性癫痫 (SMEI),被归类为一种癫痫性脑病,其特征是在出生后第一年内出现长时间的癫痫发作。这些癫痫发作经常与发烧或疾病同时出现,并且经常最初被误诊为热性惊厥。DS 的准确诊断和随后的后续治疗通常会被推迟。发病时,脑电图 (EEG) 看起来正常,神经影像学未显示任何结构异常。虽然通常会达到早期发育里程碑,但在出生后第二年可能会出现退化的迹象,常伴有抽搐性癫痫持续状态、交替性半抽搐和肌阵挛性癫痫发作。目前已可以进行基因检测,通过识别 SCN1A 基因突变来确诊。及时识别和诊断唐氏综合征,并实施适当的抗惊厥药物和综合治疗计划,可能有助于减少癫痫发作频率并改善长期发育结果。1 我们报告了一例 13 岁男孩的病例,该男孩表现为药物抵抗性癫痫发作和神经系统、认知和行为状态恶化。病例介绍 一名 13 岁的阿尔巴尼亚男孩因多形性药物抵抗性癫痫发作、认知和运动功能障碍被转诊至科索沃大学临床中心神经内科
Dravet 综合征:一例病例报告
主要关键词


相关文件推荐