机构名称:
¥ 1.0
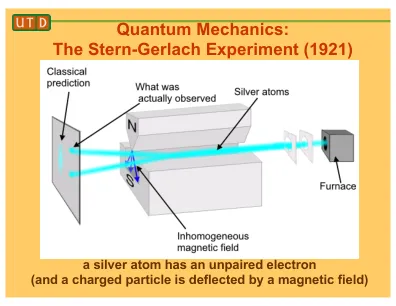
抽象的量子机械方法构成了计算化学的基石,在原子量表上提供了对分子行为和特性的前所未有的见解。这些方法阐明了通过求解Schrödinger方程来理解各种化学系统至关重要的基本电子结构,能量和性能。在这些方法中,密度功能理论(DFT)在研究原子,分子和固体的电子特性方面具有多功能性,它植根于精确的Hohenberg-kohn定理和Kohn-Sham方程。本评论探讨了计算化学中量子机械方法的宽敞景观,突出了它们在推进科学理解和技术创新方面的关键作用。许多领域,包括材料科学,催化和药物开发,利用这些技术来增强分子结构,预测反应,模拟光谱特性并澄清溶剂化效应。量子化学现在可以借助高级技术(如释放后循环方法和时间依赖性的DFT)预测更多。这些技术为我们提供了有关分子如何移动以及电子如何激发的更多信息。分子动力学(MD)模拟通过显示分子如何随时间移动和相互作用,从而增加了量子力学方法。他们通过将理论上应该与实际发生的情况联系起来来实现这一目标。关键字:量子,机械方法,计算化学。添加基于结构的药物设计(SBDD)和材料建模等计算机程序显示了量子化学如何改变事物,加快发现过程并提高分子行为的准确性。光谱模拟和溶剂化研究有助于我们预测如何解释实验数据并确定环境如何影响分子的行为及其应用,从而使计算化学更有用。量子化学软件和高性能计算框架的持续演变使对高级计算工具的访问权限,从而促进了解决复杂科学挑战的协作和创新。随着量子能力的提高,未来有望在化学和跨学科领域进行更大的应用,推动材料设计,药物开发和环境可持续性的持续进展。
计算化学中的量子力学方法
主要关键词





相关文件推荐