机构名称:
¥ 1.0
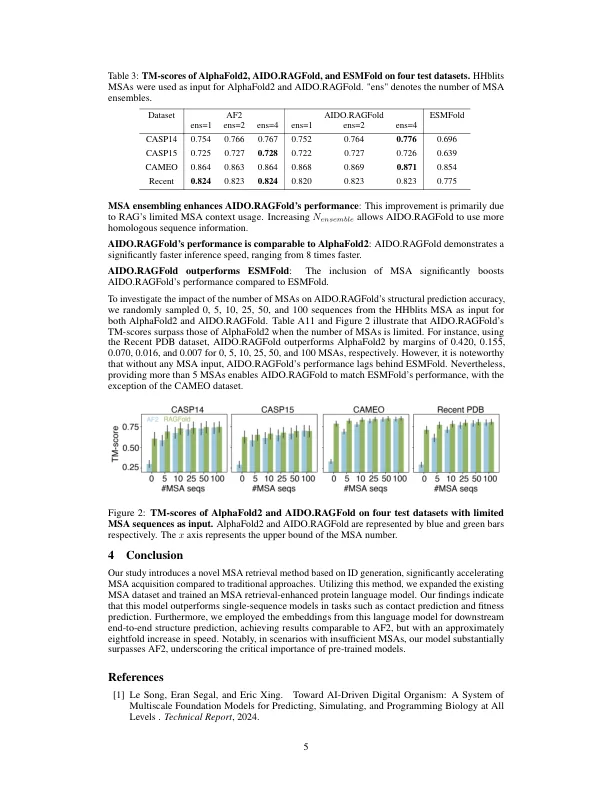
高级人工智能技术的出现在蛋白质结构预测方面取得了显着加速。alphafold2是该领域中的开创性方法,它通过利用Evoformer模块从multiple序列比对(MSA)自动提取共进化信息,为预测准确性设定了新的基准测试。但是,诸如Alphafold2之类的结构预测方法的疗效在很大程度上取决于MSA的深度和质量。为了解决这一局限性,我们提出了两个新型模型Aido.ragplm和aido.ragfold,它们是A-e-e-dected蛋白质语言模型和AI-Drigity数字有机体中的概述的模块[1]。aido.ragplm将预训练的蛋白质模型与检索的MSA整合在一起,从而使共同进化信息纳入结构前字典,同时通过大规模预处理补偿了MSA信息不足。我们的方法在困惑,接触预测和适应性预测中超过了单序蛋白语言模型。我们利用aido.ragplm作为蛋白质结构预测的特征提取器,导致aido.ragfold的发展。当有足够的MSA提供时,Aido.Ragfold就可以达到与Alphafold2相当的TM分数,并且最多运行速度长达八倍。在MSA不足的情况下,我们的方法显着优于Al-PhaFold2(∆ TM得分= 0.379、0.116和0.116和0.059,对于0、5和10 MSA序列作为输入)。我们的发现表明aido.ragplm为蛋白质结构预测提供了有效,准确的解决方案。此外,我们使用层次ID生成开发了一种从Uniclust30数据库搜索的MSA检索器,该数据库比传统方法快45至90倍,并用于扩展aido.ragplm的MSA培训集,增长32%。
通过注意蛋白质和基因组基础模型来理解蛋白-DNA相互作用
主要关键词




相关文件推荐