机构名称:
¥ 1.0
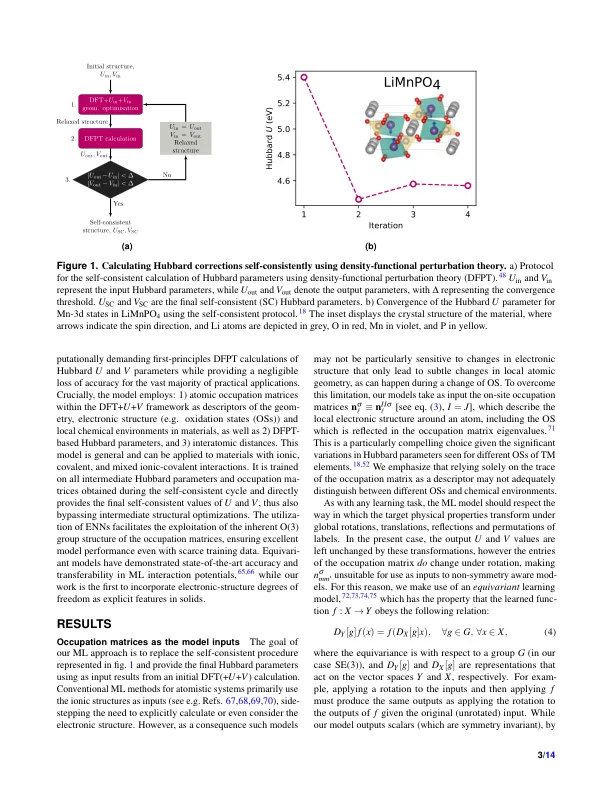
具有扩展Hubbard功能(DFT + U + V)的密度功能理论提供了一个可靠的框架,可以准确描述包含过渡金属或稀有元素的复杂材料。它是通过减轻半本地功能固有的自我相互作用误差来做到的,该误差在具有部分填充D和F电子状态的系统中特别明显。但是,在这种方法中实现准确性取决于现场U和现场v哈伯德参数的准确确定。在实践中,这些是通过半经验调整,需要先验知识或更正确地通过使用预测但昂贵的第一原理计算来获得的。在这里,我们提出了一种基于模棱两可的神经网络的机器学习模型,该模型使用原子占用矩阵作为描述符,直接捕获了手头系统的电子结构,局部化学环境和氧化状态。我们在这里以迭代性线性响应计算为单位计算的哈伯德参数的预测,如密度功能性扰动理论(DFPT)和结构放松。值得注意的是,当对跨越各种晶体结构和组成的12个材料的数据进行培训时,我们的模型分别达到了Hubbard U和V参数的平均相对误差,分别为3%和5%。通过规避计算昂贵的DFT或DFPT自洽协议,我们的模型可以显着加快用可忽略的计算开销的哈伯德参数的预测,同时接近DFPT的准确性。此外,由于其可靠性的可传递性,该模型通过高通量计算促进了加速的材料发现和设计,与各种技术应用相关。
用epivariant神经网络的机器学习哈伯德参数
主要关键词




相关文件推荐